mRNA vaccines and immune response
XU Zhiyi, 1 May 2026
Since the onset of the COVID-19 pandemic, mRNA vaccines have emerged as a highly promising approach to disease prevention and treatment. Whether targeting infectious diseases or cancer through neoantigen vaccines, they have become a key focus of corporate R&D efforts.
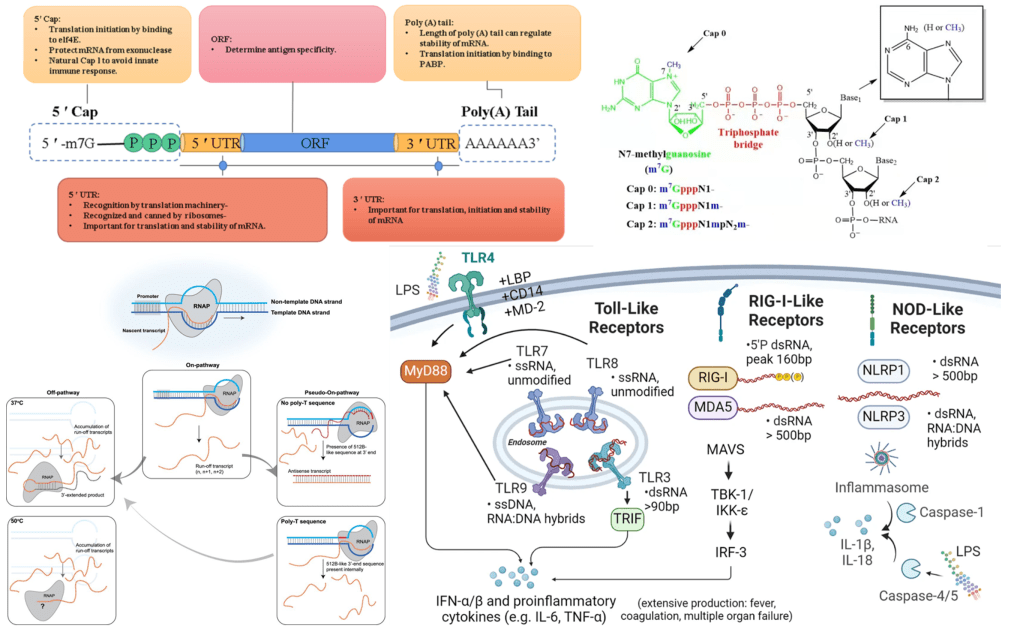
Figure 1. Therapeutic mRNA structure and immune response induced by mRNA in vivo
Currently, most of the mRNA used in pharmaceuticals is produced through in vitro transcription. Mature mRNA consists of a 5′-cap, a 5′-untranslated region (5′ UTR), an open reading frame (ORF), a 3′-untranslated region (3′ UTR), and a polyA tail. In eukaryotes, the 5′-end of mRNA is composed of m7GpppNm (cap1) or m7GpppN1mN2m (cap2), where Nm represents 2′-O-methylated nucleotides. The lack of methylation in cap0 (m7GpppN) may also lead to RIG-I activation, whereas the use of cap1 or cap2 reduces cytokine induction via the RNA sensors RIG-I and MDA5, thereby enhancing safety. Furthermore, although the known translation inhibitor interferon (IFN)-induced tripeptide repeat protein (IFIT1) competes with eIF4E for binding to cap0, its affinity for cap1 and cap2 is significantly lower. During in vitro transcription of mRNA, byproducts of dsRNA may be generated because the T7 transcriptase may not detach from the DNA template when transcribing to the 3’ end, or may mistakenly use the transcribed RNA as a template for transcription. In both immune and non-immune cells, dsRNA is recognized by RLRs and NLRs, and the signal is transmitted to the mitochondrial antiviral signaling protein (MAVS), thereby activating TBK1 and IKKξ. dsRNA can also be recognized by two members of the NOD-like receptor family (NLRs), NLRP1 or NLRP3, which promote the proteolytic maturation of the cytokines IL-1β and IL-18, trigger inflammation, and lead to highly inflammatory apoptosis. Reducing the dsRNA content in mRNA products is critical for lowering the immunogenicity of mRNA; typically, the dsRNA content should be below 0.1%-0.5%. Even trace amounts of dsRNA remaining in mRNA products can still elicit a certain immune response; therefore, some researchers view these trace amounts of dsRNA as natural adjuvants in mRNA vaccines, helping them induce a stronger immune response in the body.
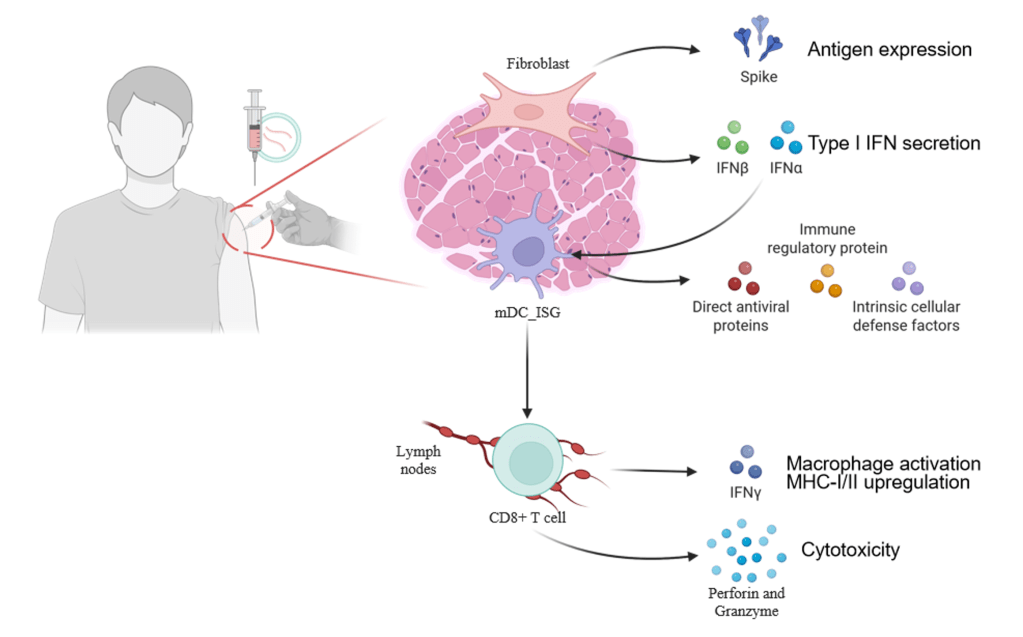
Figure 2. Immune response induced by mRNA vaccine after intramuscular injection
Studies have shown that following intramuscular administration of an mRNA vaccine, the majority of antigenic proteins are expressed and secreted by fibroblasts at the injection site. Among these fibroblasts, those highly expressing Cxcl5 and Ccl19 become dominant 16 hours after administration of the LNP-mRNA vaccine. These cells are strongly associated with cytokine-mediated signaling pathways, can present antigens via MHC-I, and induce the expression of type I IFN-related cytokines, like IL-1β. Concurrently, mRNA-specific type I IFN responses also occur in mDCs at the injection site. These mDCs subsequently migrate to nearby lymph nodes, where they activate T cells via MHC-antigen complexes to trigger subsequent immune responses.
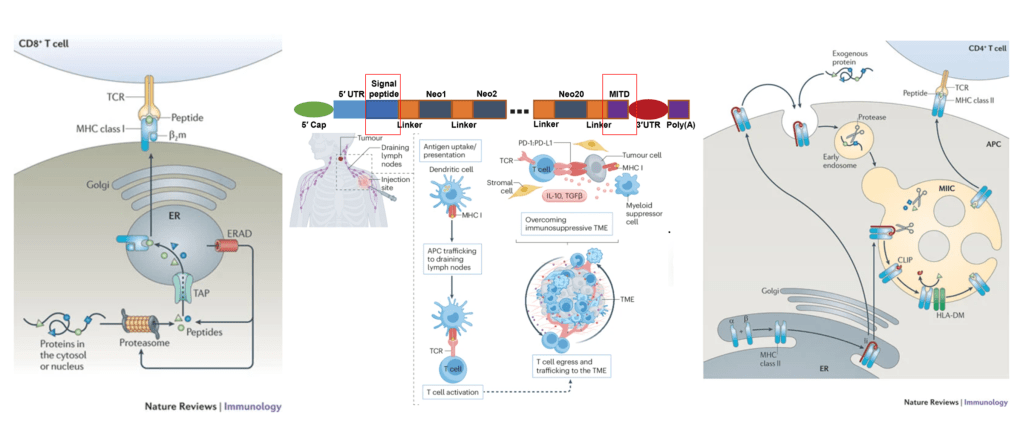
Figure 3. Immune response induced by neo-antigen mRNA vaccine
In addition to preventive vaccines for infectious diseases, the development of neoantigen vaccines for cancer treatment is also crucial. When designing neoantigen mRNA vaccines, whole-exome, RNA, or protein sequencing of patient tumor samples is typically performed. Through analysis and AI modeling, neoantigen sequences are identified that exhibit high heterogeneity, are widely distributed within the tumor, bind tightly to MHC, and have high TCR activation potency. In mRNA vaccines, typically a dozen to several dozen neoantigen sequences are concatenated, with a secretory signal peptide added to the 5’ end and a major histocompatibility complex (MHC) class I transport signal peptide added to the 3’ end. This design ensures that the neoantigens can be presented by MHC-I intracellularly and, after secretion into the extracellular space, presented by antigen-presenting cells (APCs) via MHC-II. Neoantigen mRNA vaccines can typically rapidly activate CD4+ and CD8+ T cells and mobilize CD8+ T cells to effectively kill tumors in vivo. At the same time, prior administration of neoantigen vaccines can also enhance the body’s immune response to tumor cells expressing these antigens, thereby preventing tumor growth.
Reference
- Vlatkovic, I. Non-Immunotherapy Application of LNP-MRNA: Maximizing Efficacy and Safety. Biomedicines 2021, 9 (5), 530. https://doi.org/10.3390/biomedicines9050530.
- Zhang, J.; Liu, Y.; Li, C.; Xiao, Q.; Zhang, D.; Chen, Y.; Rosenecker, J.; Ding, X.; Guan, S. Recent Advances and Innovations in the Preparation and Purification of in Vitro-Transcribed-MRNA-Based Molecules. Pharmaceutics 2023, 15 (9), 2182. https://doi.org/10.3390/pharmaceutics15092182.
- Li, Y.; Wang, M.; Peng, X.; Yang, Y.; Chen, Q.; Liu, J.; She, Q.-B.; Tan, J.; Lou, C. D.; Liao, Z.; Li, X. MRNA Vaccine in Cancer Therapy: Current Advance and Future Outlook. Clinical and translational medicine 2023, 13 (8). https://doi.org/10.1002/ctm2.1384.
- Kim, S.; Jeon, J. H.; Kim, M.; Lee, Y.; Hwang, Y.-H.; Park, M.; Li, C. H.; Lee, T.; Lee, J.-A.; Kim, Y.-M.; Kim, D.; Lee, H.; Kim, Y.-J.; Kim, V. N.; Park, J.-E.; Yeo, J. Innate Immune Responses against MRNA Vaccine Promote Cellular Immunity through IFN-β at the Injection Site. Nature Communications 2024, 15 (1). https://doi.org/10.1038/s41467-024-51411-9.
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising Targets for Cancer Therapy. Signal Transduction and Targeted Therapy 2023, 8 (1). https://doi.org/10.1038/s41392-022-01270-x.
- Saxena, M.; van der Burg, S. H.; Melief, C. J. M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nature Reviews Cancer 2021, 21 (6), 360–378. https://doi.org/10.1038/s41568-021-00346-0.
Artificial Intelligence in Immunology: From Immune Prediction to Therapeutic Design
Rong Yiou, 22 Feb 2026
The immune system is one of the most complex biological systems in nature, characterized by enormous molecular diversity, dynamic cellular interactions, and rapid adaptation to pathogens. Advances in high-throughput technologies such as single-cell RNA sequencing (scRNA-seq), TCR/BCR repertoire sequencing, proteomics, cryo-electron microscopy, and structural biology have generated unprecedented amounts of immunological data. However, traditional computational approaches often struggle to interpret these highly dimensional datasets. Artificial intelligence (AI), particularly machine learning (ML) and deep learning (DL), has emerged as a transformative tool that enables researchers to identify hidden patterns in immune datasets, predict immune responses, design therapeutics, and accelerate immunological discovery (Figure 1). AI is increasingly being integrated into cancer immunotherapy, infectious disease research, autoimmune disease studies, vaccine development, and antibody engineering.
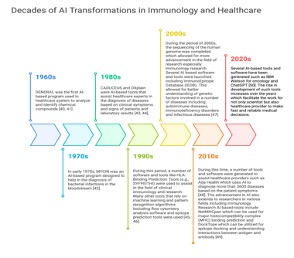
Figure 1: decades of AI transformations in Immunology and Healthcare
One of the earliest and most impactful applications of machine learning in immunology involves analyzing adaptive immune receptor repertoires. T-cell receptors (TCRs) and B-cell receptors (BCRs) are generated through V(D)J recombination, creating extraordinary diversity that enables adaptive immunity. A single individual may possess approximately 10⁷–10⁸ unique TCRs, while the theoretical diversity of possible receptors exceeds 10¹⁵ sequences. These receptors contain highly variable complementarity-determining region 3 (CDR3) sequences, which largely determine antigen specificity. Immune repertoire sequencing typically begins with raw sequencing reads that are processed through V(D)J alignment (Figure 2) and clonotype identification using tools such as MiXCR, IgBLAST, and the IMGT platform. Traditional machine learning algorithms such as Random Forests, Support Vector Machines, and k-nearest neighbors were initially applied to classify disease-associated immune repertoires. More specialized immunology-focused tools such as GLIPH cluster TCRs based on shared antigen-recognition motifs, while TCRdist measures functional similarity between receptors. These approaches have been used to identify infection-associated immune signatures, characterize vaccine responses, and detect tumor-reactive T cells.
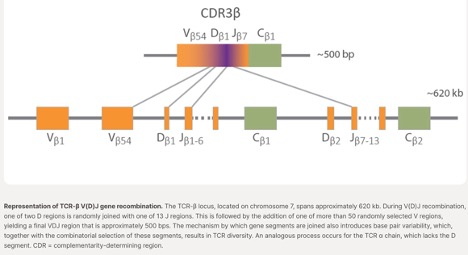
Figure 2. Fundamentals of VDJ alignment on which algorithms are based.
Another major challenge in immunology is predicting whether a specific T-cell receptor recognizes a specific antigen. T-cell activation requires highly specific interactions between TCR α chains, TCR β chains, peptide antigens, and major histocompatibility complex (MHC) molecules (Figure 3). Even minor sequence variations can dramatically alter recognition outcomes. Deep learning models such as NetTCR were developed to address this problem by learning sequence patterns associated with antigen binding. NetTCR uses one-hot encoded TCR and peptide sequences as input, followed by convolutional neural networks (CNNs) that automatically identify sequence motifs involved in recognition (Figure 4). In a landmark study, Montemurro et al. demonstrated in Nature that NetTCR-2.0 significantly improved prediction accuracy by incorporating paired TCR α/β sequence information from databases such as VDJdb, McPAS-TCR, and the Immune Epitope Database. Their model showed that paired receptor information substantially improved antigen specificity prediction and could accelerate adoptive T-cell therapy development and personalized cancer immunotherapy.[1]
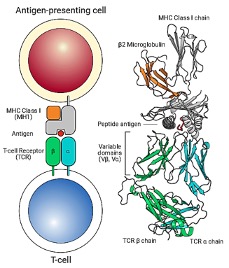
Figure 3. T cells recognize antigens through interaction between: T-cell receptor (TCR) (alpha and beta chain), peptide, and MHC molecule. This complex is called pMHC (peptide–MHC complex).
While NetTCR focuses on individual receptor-antigen interactions, DeepTCR analyzes entire T-cell repertoires. DeepTCR uses embedding layers, convolutional neural networks, and variational autoencoders (VAEs) to identify latent patterns in large immune datasets. Sidhom et al. demonstrated in Nature Communications that DeepTCR could identify antigen-specific T-cell populations from highly noisy datasets generated from tetramer sorting, single-cell sequencing, and tumor samples.[2] The model successfully identified influenza-specific T cells, Epstein-Barr virus-reactive T cells, and melanoma-associated immune clones, demonstrating how AI can uncover therapeutically relevant immune populations that may otherwise remain hidden within massive datasets.
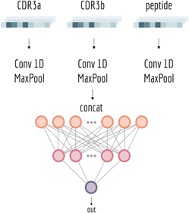
Figure 4. Simplified NetTCR model.
Beyond immune receptor prediction, AI has become indispensable for analyzing single-cell immune profiling datasets. Single-cell sequencing has revolutionized immunology by enabling researchers to study individual immune cells rather than bulk populations. However, datasets often contain millions of cells and thousands of genes per cell. Machine learning algorithms are required for clustering immune populations, identifying cell states, performing trajectory inference, and analyzing cell-cell communication networks. Tools such as Seurat, Scanpy, Harmony, and CellChat have become foundational in modern tumor immunology, autoimmune disease research, and infectious disease studies.
Immune recognition is also fundamentally a structural biology problem, making protein structure prediction another major AI application in immunology. Antibody-antigen binding, TCR-peptide-MHC interactions, and cytokine receptor signaling all depend on precise three-dimensional molecular interactions. The development of AlphaFold by DeepMind revolutionized structural biology by predicting highly accurate protein structures directly from amino acid sequences (Figure 5). AlphaFold first performs multiple sequence alignment using databases such as UniRef and MGnify, identifies co-evolving residues, constructs pairwise residue maps, processes these representations through its transformer-based Evoformer architecture, and finally predicts three-dimensional atomic coordinates with confidence scores such as pLDDT.[3] During the COVID-19 pandemic, AlphaFold-enabled structural predictions accelerated analysis of the SARS-CoV-2 spike protein, helping researchers identify receptor-binding domains, antibody epitopes, and immune escape mutations that informed vaccine development.
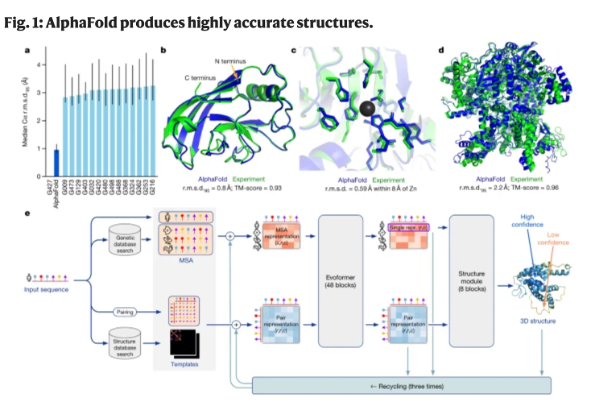
Figure 5. The development of AlphaFold in predicting 3D protein structure
AI is also transforming therapeutic antibody development. Therapeutic antibodies such as Pembrolizumab and Trastuzumab have revolutionized treatment for cancer and autoimmune diseases, but traditional antibody discovery methods remain expensive and slow. Deep learning models such as DeepAb predict antibody structures directly from sequence by using bidirectional LSTM networks to model heavy and light chain interactions and predict inter-residue geometry constraints that reconstruct antibody structures.[4] This is especially useful for modeling highly variable CDR-H3 loops that are critical for antigen recognition. In parallel, transformer-based protein language models such as ESM, ProtBERT, and AntiBERTy treat antibody sequences as biological language and learn sequence-function relationships from millions of antibodies. These models can predict mutation effects, optimize binding affinity, and even generate entirely novel antibody candidates.
The future of AI in immunology will likely involve multimodal learning systems that integrate single-cell sequencing, proteomics, structural biology, imaging, and clinical data to create comprehensive models of immune behavior. AI-driven personalized immunotherapy may predict patient-specific immune responses, while generative biology approaches may design entirely novel immune molecules. Ultimately, the integration of artificial intelligence with experimental immunology is shifting the field from descriptive observation toward predictive and design-driven science, creating new opportunities for precision medicine, immunotherapy, and next-generation vaccines.
Reference
- Montemurro, A., Schuster, V., Povlsen, H.R. et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun Biol 4, 1060 (2021). https://doi.org/10.1038/s42003-021-02610-3Sidhom JW, et al. Nat Commun. 2021;12:1605.
- Sidhom, JW., Larman, H.B., Pardoll, D.M. et al. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat Commun 12, 1605 (2021). https://doi.org/10.1038/s41467-021-21879-wRuffolo JA, et al. Bioinformatics. 2021;37(13):2127–2129.
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
- Mason, D.M., Friedensohn, S., Weber, C.R. et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat Biomed Eng 5, 600–612 (2021). https://doi.org/10.1038/s41551-021-00699-9
Reprogramming tumor-associated macrophages: mechanisms and therapeutic strategies for cancer immunotherapy
LI Xiaojing, 5 Feb 2026
Macrophages are innate immune cells with the plasticity of phenotype, existing in a spectrum of functional states that profoundly influence tumor progression. Macrophages are broadly classified into classically activated (M1) and alternatively activated (M2) phenotypes (Figure 1). M1 macrophages, induced by signals like IFN-γ and LPS, exhibit pro-inflammatory, antimicrobial, and antitumoral activities. In contrast, M2 macrophages, stimulated by cytokines such as IL-4, IL-13, and TGF-β, are associated with anti-inflammatory responses, tissue repair, and immunosuppression. M2 macrophages are further subdivided into M2a, M2b, M2c, and M2d, each with distinct markers, cytokines, and roles in promoting tumor progression, angiogenesis, and immune evasion.
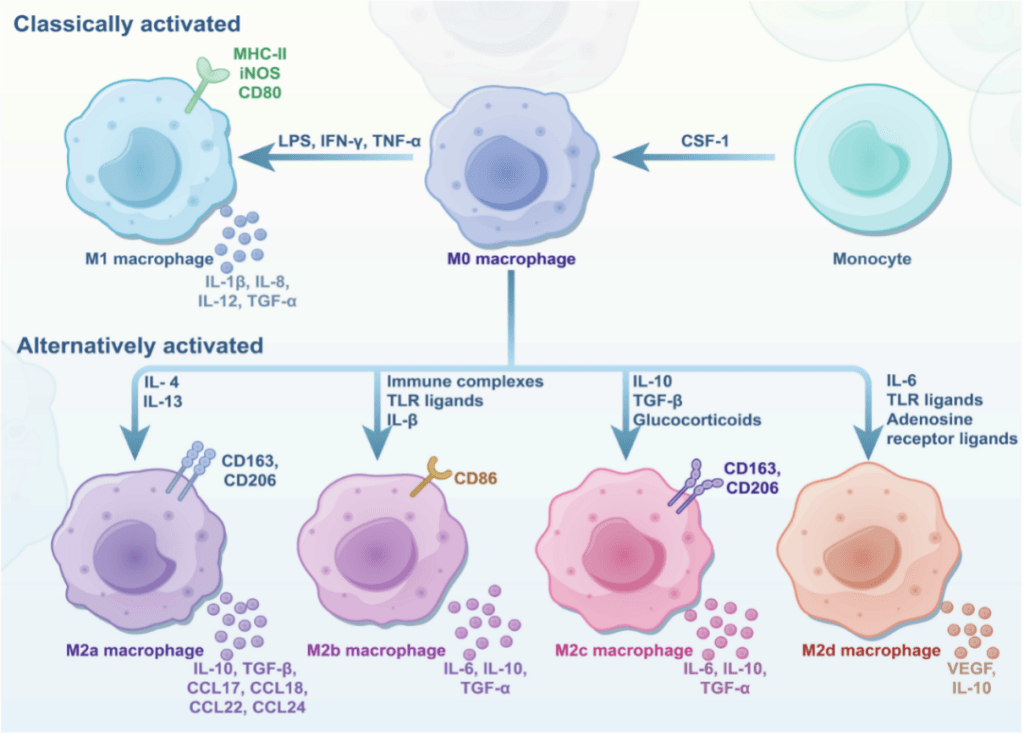
Figure 1. The polarization of macrophages into classically activated (M1) and alternatively activated (M2) phenotypes.[1]
The polarization of macrophage into M1 or M2 is a precisely regulated process comprising several key regulatory networks. Figure 2 demonstrates numerous strategies essential for macrophage polarization and depicts feedback control on signaling pathways of M1 and M2. M1 activation mainly relies on TLR4-NF-κB and IFN-γ-STAT1 pathways, while M2 polarization is mediated by IL-4/13-STAT6 and IL-10-STAT3 signaling, with negative regulators such as SOCS3 fine-tuning these responses. Also, macrophage polarization can be induced by GO (graphene oxide) towards the M1 phenotype. Nanoparticles, such as HA-PEI/pDNA-IL-10 NPs and alginate NPs containing cytokine IL-10 plasmid DNA, can modulate programming from M1 toward M2.
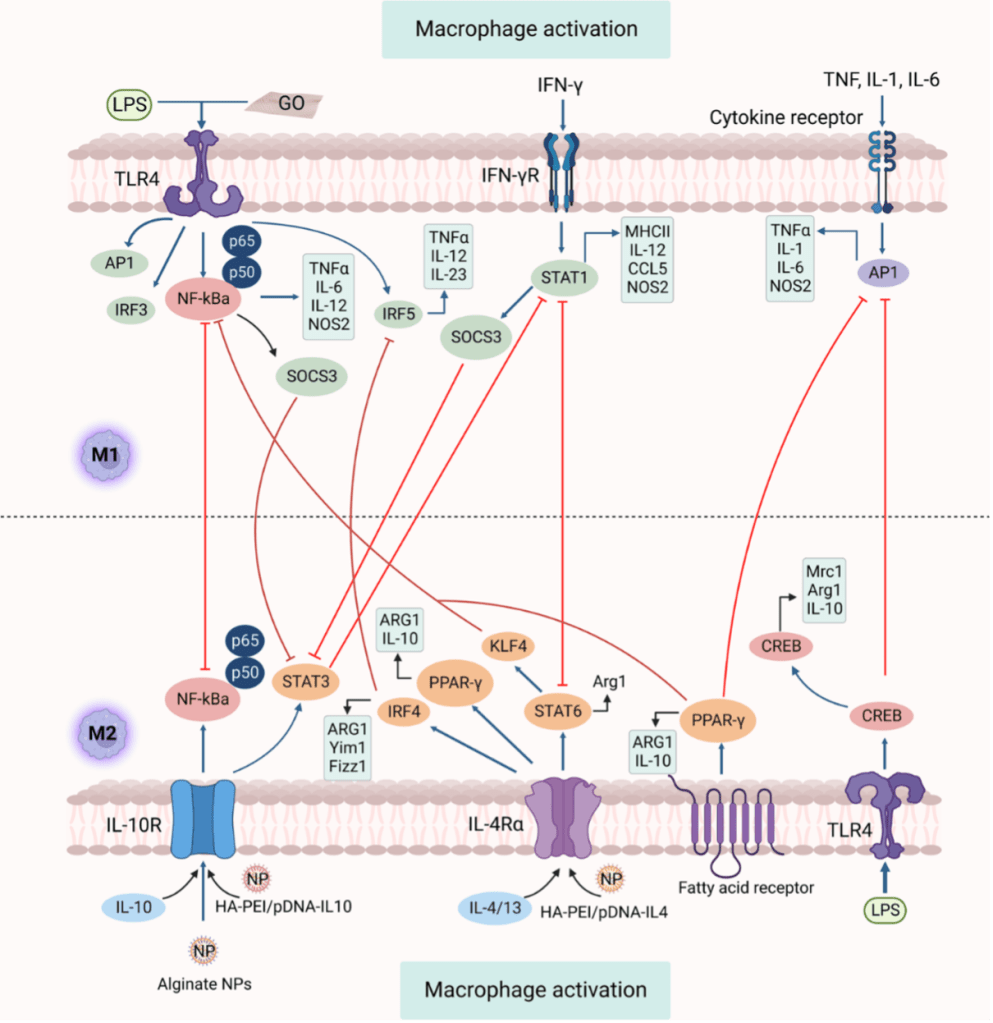
Figure 2. Pathways for signaling macrophage polarization.[2]
In the tumor microenvironment (TME), tumor-associated macrophages (TAMs) predominantly exist as M2-like cells and promote tumor cell proliferation, angiogenesis, and the formation of an immunosuppressive microenvironment that facilitates tumor metastasis (Figure 3). In addition to undergoing polarization and repolarization in response to microenvironmental signals, TAMs influence the TME through the expression of specific surface markers and the secretion of growth factors and other bioactive molecules. CD24 and CD47 on tumor cells bind to Siglec-10 and SIRPα on M2 TAMs, enabling tumor cells to evade macrophage phagocytosis through the “don’t eat me” pathway. M2 TAMs promote tumor migration and invasion by secreting MMP-12 and cathepsins. They also inhibit inflammatory cell function through anti-inflammatory mediators (IL-10, TGF-β), creating an immunosuppressive TME. Additionally, M2 TAMs support tumor angiogenesis and by secreting PDGF, VEGFA, and EGF. In contrast, M1 TAMs release proinflammatory factors that counteract tumor progression.
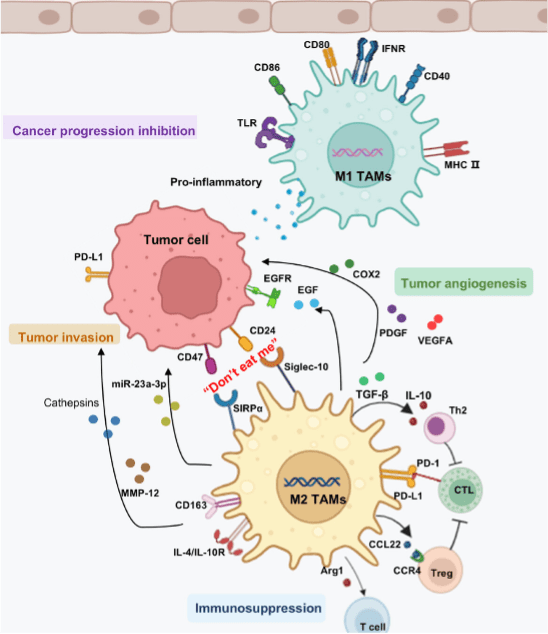
Figure 3. Effects of tumor-associated macrophages (TAMs) on tumor progression within the tumor microenvironment.[3]
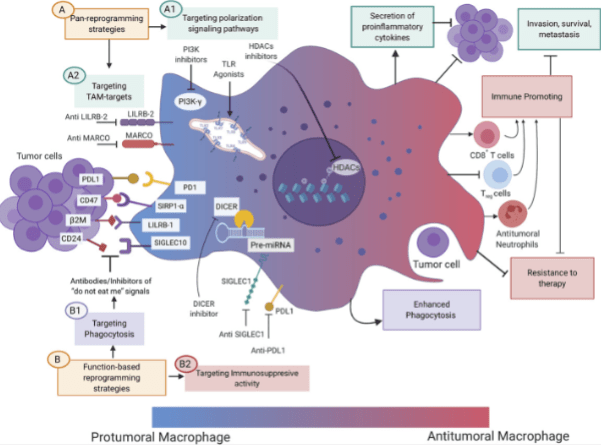
Figure 4. Therapeutic strategies aimed at changing the phenotype of TAMs from a protumoral to an antitumoral state (macrophage reprogramming).[4]
Reference
1. Guan F, Wang R, Yi Z, et al. Tissue macrophages: origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct Target Ther. 2025;10(1):93.
2. Chen S, Saeed AFUH, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8(1):207.
3. Liu Z, Li Y, Cao J, Qiu Y, Yu K, Deng S. The Role of Macrophages in Cancer: From Basic Research to Clinical Applications. MedComm. 2025;7(1):e70547.
4. Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci. 2021;1499(1):18-41.
Cancer-immunity cycle
Shen XIaoxi, 4 Dec 2025
The cancer-immune cycle describes how the body recognizes and eliminates cancer cells, functioning as a closed loop from antigen release to repeated T-cell activation. Tumor cells first release antigens as a result of mutations or abnormal protein expression. These antigens are captured and processed by dendritic cells, which present them via MHC I and MHC II molecules to CD8⁺ and CD4⁺ T cells, respectively, thereby initiating cytotoxic and helper immune responses. Dendritic cells then migrate to the lymph nodes and activate naive T cells through three essential signals: TCR recognition of antigen–MHC complexes, co-stimulatory signaling, and the cytokine environment. During this stage, CTLA-4 functions as an early “brake” by inhibiting excessive activation. Once activated, effector T cells proliferate and enter circulation, migrating precisely to the tumor through selectin-mediated rolling, chemokine-triggered integrin activation, and firm adhesion to ICAM-1/VCAM-1 on endothelial cells. After infiltrating the tumor tissue, effector T cells recognize tumor-presented antigens and execute killing through multiple mechanisms, including perforin/granzyme release, Fas–FasL signaling, and secretion of IFN-γ. This process induces tumor cell apoptosis and simultaneously releases new antigens, further amplifying the immune cycle through “antigen spreading.”
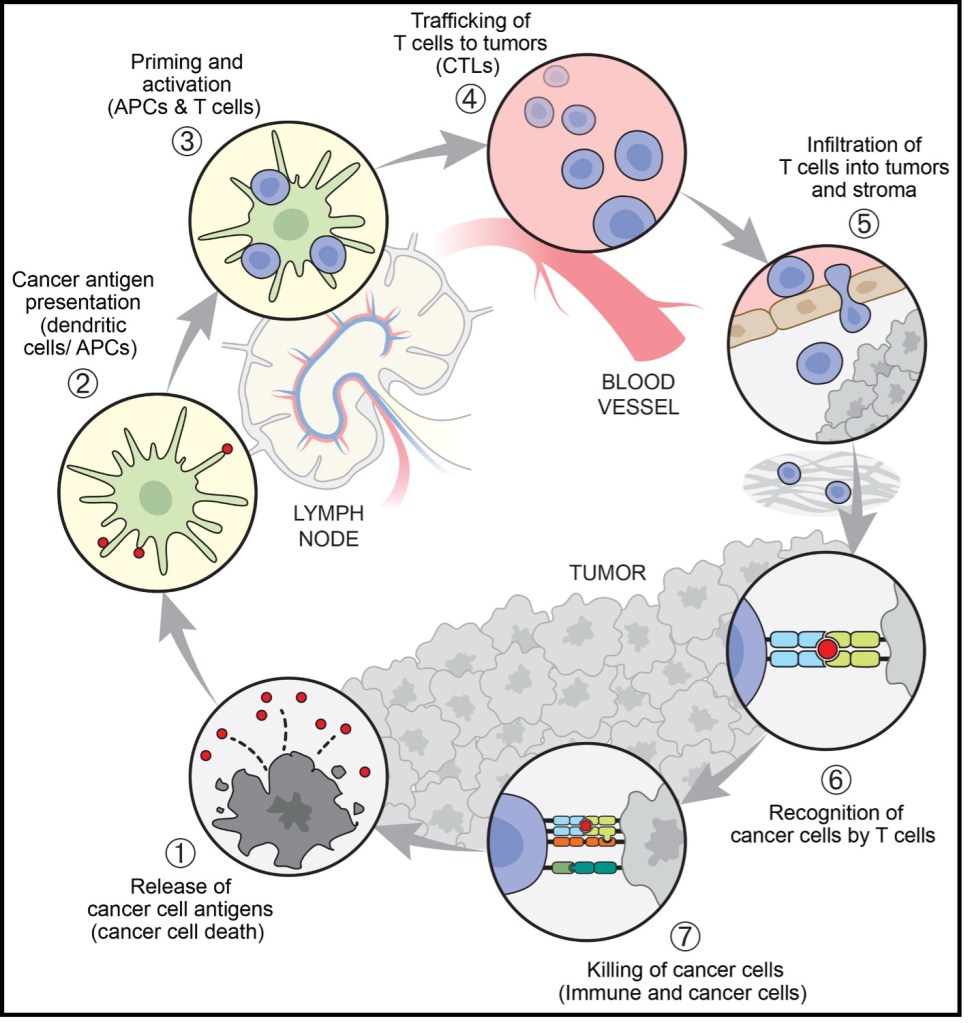
Figure 1. The seven steps of the cancer-immunity cycle.
However, most tumors interrupt this cycle at various stages, creating immune-evasive barriers that result in three immune phenotypes, among which the “immune-desert” and “immune-excluded” types are considered “cold tumors.” Immune-desert tumors often arise from insufficient antigen release or impaired antigen presentation, preventing T-cell priming. Immune-excluded tumors are characterized by T cells trapped in the stromal periphery due to abnormal vasculature, lack of chemokines, or dense extracellular matrix. Even when T cells infiltrate the tumor, the microenvironment often suppresses their activity through immunosuppressive cells and molecules such as IL-10, TGF-β, IDO, adenosine, as well as hypoxia and acidic metabolism. Additionally, high PD-L1 expression on tumor cells binding to PD-1 on T cells induces functional exhaustion.
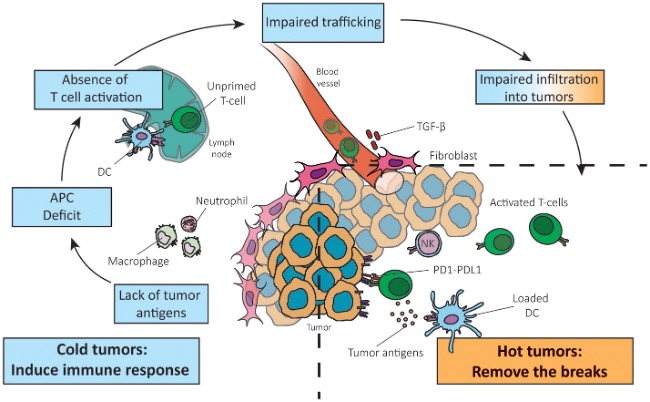
Figure 2. Reversing a cold into a hot tumor, and which step of the anti-cancer immune response is not functional in cancers.
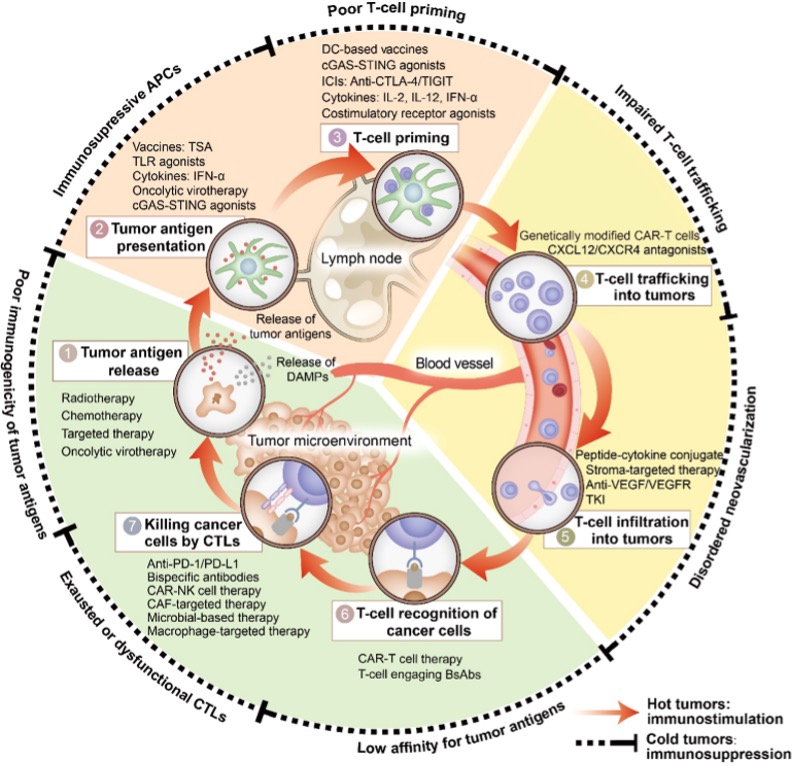
Figure 3. Immune-based combination therapies convert cold tumors into hot tumors by regulating the cancer-immunity cycle.
Based on these barriers, modern cancer immunotherapy aims to convert “cold tumors” into “hot tumors.” Upstream interventions such as radiotherapy, chemotherapy, oncolytic viruses, tumor vaccines, STING agonists, and HDAC inhibitors enhance antigen release and dendritic cell function. During the trafficking stage, anti-VEGF therapies can normalize vasculature and restore chemokine signaling, while engineered CAR-T/TCR-T cells may improve tumor homing. Within the tumor microenvironment, immune checkpoint inhibitors targeting PD-1/PD-L1 or CTLA-4 release T cells from suppression, and inhibitors of IDO or adenosine pathways, as well as strategies to reprogram tumor-associated macrophages or deplete MDSCs, can further restore immune activity. Given the complexity of immune evasion, combination therapies—such as radiotherapy plus checkpoint inhibitors, anti-angiogenic therapy plus immunotherapy, or simultaneous targeting of multiple suppressive pathways—have become a major trend, aiming to break through multiple immune barriers and drive the tumor immune cycle into a sustained, self-reinforcing anti-tumor response.
Reference
- Mellman, I., Chen, D. S., Powles, T., & Turley, S. J. (2023). The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity, 56(10), 2188-2205.
- Bonaventura, P., Shekarian, T., Alcazer, V., Valladeau-Guilemond, J., Valsesia-Wittmann, S., Amigorena, S., … & Depil, S. (2019). Cold tumors: a therapeutic challenge for immunotherapy. Frontiers in immunology, 10, 168.
- Ni, J. J., Zhang, Z. Z., Ge, M. J., Chen, J. Y., & Zhuo, W. (2023). Immune-based combination therapy to convert immunologically cold tumors into hot tumors: an update and new insights. Acta Pharmacologica Sinica, 44(2), 288-307.
- Abbas, A. K., Lichtman, A. H., & Pillai, S. (2019). Basic immunology: Functions and disorders of the immune system, 6e: Sae-E-Book. Elsevier Health Sciences.
T cell exhaustion
YUAN Dingdong, 11 Nov 2025
T cell exhaustion refers to a state of dysfunction exhibited by CD8+ T cells under chronic antigen stimulation. It mainly occurs in chronic viral infections and cancer. Exhausted T cells demonstrate reduced production of partial cytokines, weakened cytotoxicity, high expression of inhibitory receptors, decreased or completely lost proliferative ability, and eventually apoptosis.
T cell exhaustion results from the combined action of multiple factors (Figure 1), mainly including: 1. Persistent antigen stimulation, which is the main driver of T cell exhaustion; 2. Tumor microenvironment, where immunosuppressive factors such as IL10 and TGFβ promote T cell exhaustion. Additionally, hypoxia and nutrient deprivation further exacerbate T cell exhaustion; 3. Intercellular interactions, regulatory cells like Tregs play an important role in T cell exhaustion. Beyond Tregs, other regulatory cell types like immunoregulatory APCs, MDSCs, NK cells, and even CD8+ regulatory populations may influence viral persistence in chronic infections, directly or indirectly promoting T cell exhaustion. 4. High expression of inhibitory receptors such as PD1, TIM3, CTLA-4, and LAG3.
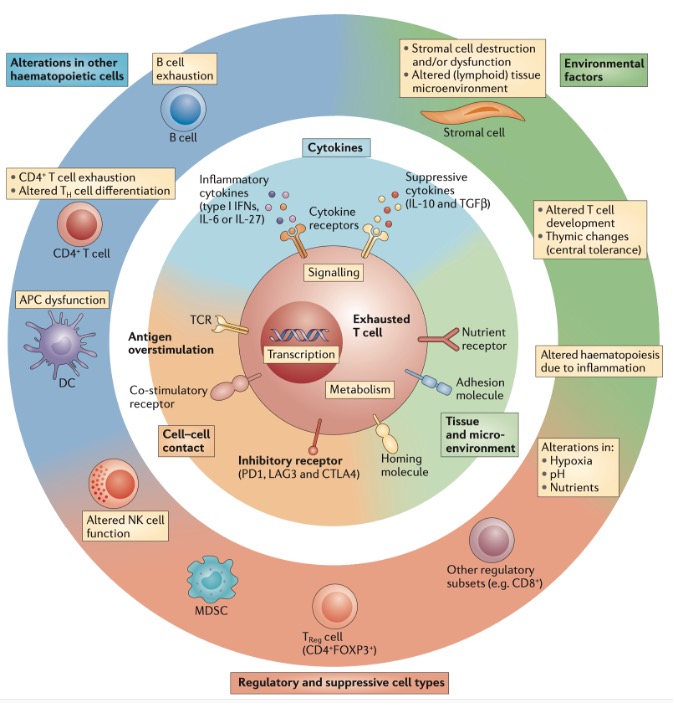
Figure 1. Overview of mechanisms of T cell exhaustion.
Inhibitory receptors regulate T cell exhaustion by influencing T cell molecular signaling pathways. Typically, T cell activation requires three conditions: binding of TCR to the MHC antigen complex on APCs; 2. Co-stimulatory signals from CD28 and CD80/CD86 on APCs; and cytokine IL2. The inhibitory receptor PD1, possessing an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) in its intracellular region, recruits SHP2 to inhibit CD28 and TCR signaling upon binding to PDL1 or PDL2. Another inhibitory receptor, CTLA-4, competitively binds to CD80/CD86 with CD28 to suppress CD28 signaling. CTLA-4’s intracellular region contains a tyrosine inhibitory motif YVKM, which is phosphorylated after binding to CD80/CD86, recruiting SHP2 to inhibit TCR signaling. Other inhibitory receptors, such as LAG3 and TIM3, regulate T cell exhaustion by suppressing TCR or CD28 signaling through various mechanisms.
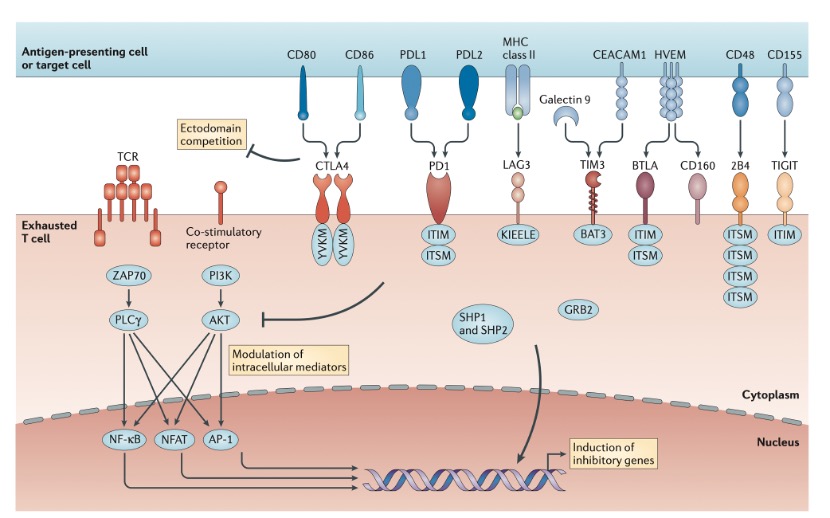
Figure 2. Molecular pathways of inhibitory receptors associated with T cell exhaustion.

Figure 3. Overview of strategies to mitigate or interrupt CAR T cell exhaustion
With a deeper understanding of T cell exhaustion, efforts have begun to reverse or mitigate it. The first approach is reversing T cell exhaustion by blocking immune inhibitory receptors, such as monoclonal antibody drugs targeting inhibitory receptors, including anti-PD1 antibody, anti-CTLA-4 antibody, and anti-LAG3 antibody. Furthermore, in clinical studies, the combination of inhibitory receptor monoclonal antibodies with other inhibitors, like cytokine inhibitors (e.g., IL10), has shown to enhance the suppression of T cell exhaustion. Another approach involves modulating the microenvironment to address exhaustion metabolically. In the tumor microenvironment where hypoxia disrupts T cell mitochondrial metabolism. Studies suggest overexpression of PGC-1α can enhance mitochondrial function in T cells, mitigating exhaustion, or the addition of acetylated cysteine to reduce ROS levels can reverse T cell exhaustion. Beyond traditional T cell therapy, exhaustion is also observed in CAR-T therapy. Addressing CAR-T exhaustion utilizes approaches (Figure 3) such as 1. Increasing CAR T cell to tumor cell ratios; Optimizing T cell culture conditions (reduce the duration of culture time, PI3K/AKT inhibition and cytokine supplementation); 3. CAR engineering to optimize signaling (Inhibit CD3 phosphorylation and mutate CD28 co-stimulatory domains); 4. Engineering transcriptional programs (Disrupt transcriptional program); 5. Mitigating metabolic exhaustion to mitigate or resolve exhaustion issues in CAR-T cells.
In summary, T cell exhaustion is not ‘bad’ T cells but rather a stalemated state between T cells and antigens under prolonged exposure. T cell exhaustion is a complex biological phenomenon involving multiple regulatory mechanisms. Understanding its mechanisms and heterogeneity is crucial for developing more effective cancer immunotherapy strategies (Figure 5). Future research will focus on how to effectively reverse the exhausted state to enhance therapeutic efficacy and reduce side effects.
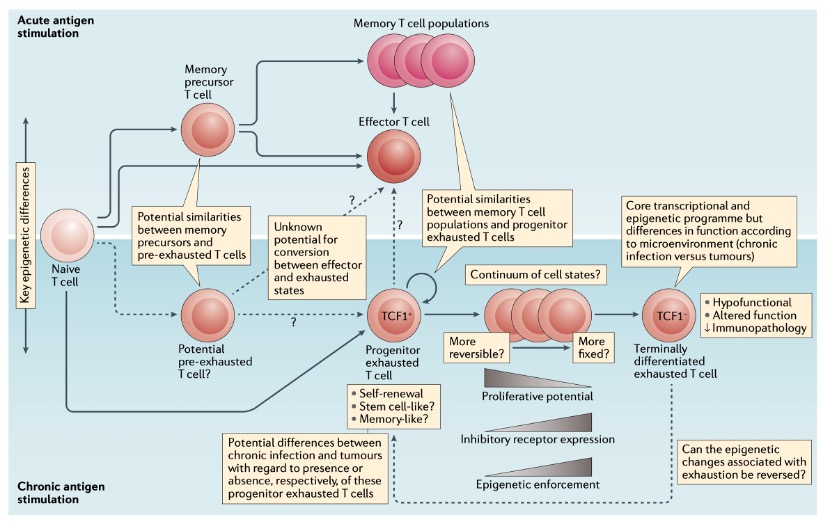
Figure 4. The knowns and unknowns of T cell exhaustion.
Reference
- Wherry, E. J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nature Reviews Immunology 2015, 15 (8), 486–499. DOI: 10.1038/nri3862.
- Pauken, K. E.; Wherry, E. J. Overcoming T Cell Exhaustion in Infection and Cancer. Trends in Immunology 2015, 36 (4), 265–276. DOI: 10.1016/j.it.2015.02.008.
- Chow, A.; Perica, K.; Klebanoff, C. A.; Wolchok, J. D. Clinical Implications of T Cell Exhaustion for Cancer Immunotherapy. Nature Reviews Clinical Oncology 2022. DOI: 10.1038/s41571-022-00689-z.
Cytokines
CHANG Jingyue, 14 Oct 2025
Cytokines are small molecule proteins secreted by various cells during immune responses, mainly used for intercellular signal transmission and for regulating immune and inflammatory responses by controlling cell proliferation, differentiation, and antibody secretion by immune cells. The signal transmission of cytokines can be classified into three modes: autocrine, paracrine, or endocrine (Figure 1).
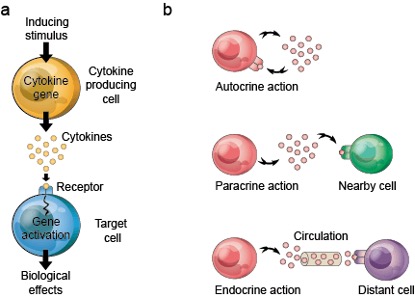
Figure 1. Signal transmission of cytokines.
Generally speaking, cytokines can be distinguished based on their molecular structure, source and function. In terms of structure, cytokines can be classified into hematopoietic cytokine family, interferon family, chemokine family or tumor necrosis factor (TNF) family. In terms of molecular function, different structures of cytokines exhibit different functions, such as regulation of immune cells, anti-viral activity, allergic reactions, cell apoptosis and transport. At the immune level, cytokines can be classified into pro-inflammatory and anti-inflammatory factors, although some cytokines simultaneously exhibit both characteristics under different conditions. Another typical way to distinguish cytokines is through different receptors, as the receptors corresponding to different cytokines show significant differences. Figure 2 shows the structures of different typical cytokine receptors.
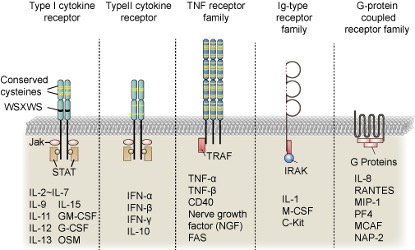
Figure 2. Classification of cytokine receptors.
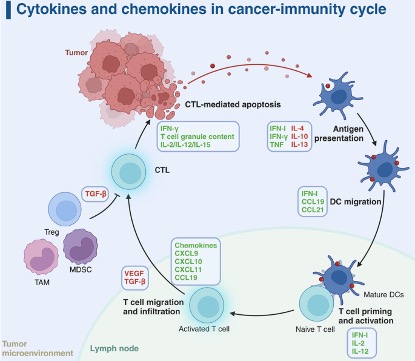
Figure 2. Classification of cytokine receptors.
The signaling pathways involved in cytokine regulation showed high complexity based on different receptors, while they share similar features in downstream signaling. The most typical signaling pathway is JAK-STAT, in which phosphorylation of tyrosine residues induce transcription and translation of target genes involved in various immune responses. Common downstream pathways include NF-κB, p38 and MAPK-related signals such as ERK and MEK, promoting cell growth, migration, survival and invasion.
In cancer immunity, cytokines exert bidirectional functions in the tumor microenvironment through pro-inflammatory and anti-inflammatory approaches (Figure 3). Major proinflammatory cytokines include IL-1 family, IL-6, and TNF, which increase inflammation, angiogenesis, ECM degradation, EMT, immunosuppressive cells, and inflammatory factors in the tumor microenvironment, thereby leading to tumor growth, proliferation, progression, metastasis, and hyperinflammation. In addition to their pro-tumor effects, these pro-inflammatory cytokines may also act as anti-tumor factors to inhibit tumor growth, proliferation, progression, metastasis and inflammatory response by increasing tumor cell apoptosis, ECM remodeling, effector immune cell function and inhibiting inflammatory factors in TME.
Existing strategies for targeting cytokines and chemokines in cancer therapy can be broadly divided into four categories. The first is to block tumorigenic cytokine signaling, including the use of antibody traps, cytokine inhibitors, antisense oligonucleotides (ASOs), and vaccines. Strategies to express antitumor cytokines or activate their downstream pathways can in turn be used, including direct cytokine or modified cytokine delivery, immune cytokine engineering, cytokine expressing cells, or cytokine delivery using viral vectors or liposomes (Figure 4). There are currently over twenty cytokine drugs approved by the FDA, covering various aspects such as tumor treatment, immune inflammation, and antiviral therapy. Its combination with other antibodies is also a popular treatment widely studied.
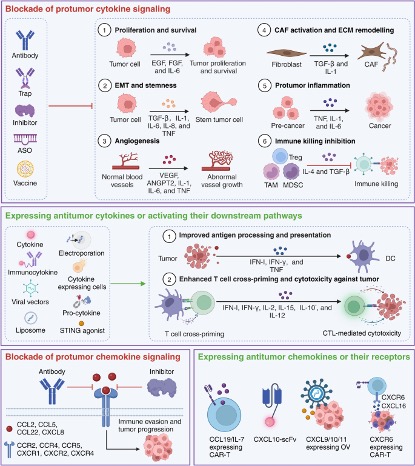
Figure 4. Clinical application of existing cytokines in cancer therapy.
Reference
- 1. Janeway. Immunobiology. Ninth Edition, CHAPTER 7 & CHAPTER 8.
- Fu Y, Tang R and Zhao X (2023) Engineering cytokines for cancer immunotherapy: a systematic review. Front. Immunol. 14:1218082.
- Yi, M., Li, T., Niu, M. et al. (2024) Targeting cytokine and chemokine signaling pathways for cancer therapy. Sig Transduct Target Ther 9, 176.
B-Cell Receptor (BCR) and Antibody (Immunoglobulin)
LI Biquan, 12 Sept 2025
B lymphocytes develop from lymphoid stem cells in the bone marrow of mammals or the bursa of Fabricius in birds, hence the name B cells. Mature B cells primarily reside in the lymphoid follicles of peripheral lymphoid organs, accounting for approximately 20% of the total peripheral lymphocyte population. B cells contribute to specific humoral immunity by producing antibodies, serve as antigen-presenting cells, and participate in immune regulation. B cells develop in the bone marrow and migrate to peripheral lymphoid organs, where antigens can activate them (Figure 1). B-lineage cell development progresses through several stages marked by the rearrangement and expression of immunoglobulin genes, as depicted in Figure 2.
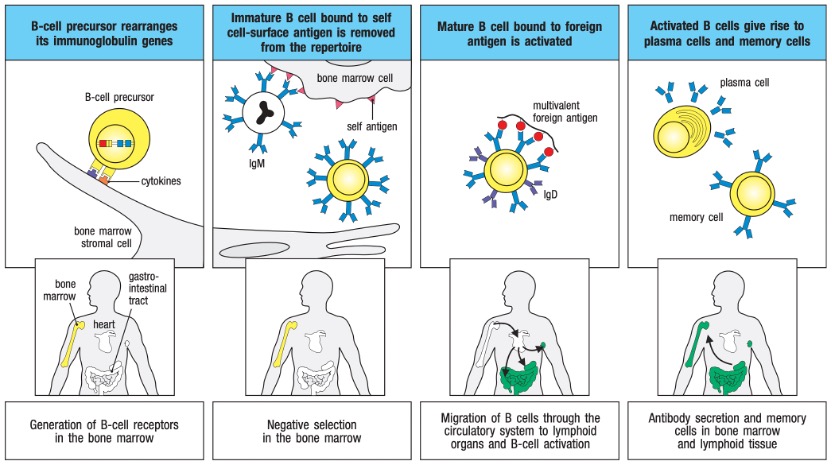
Figure 1. Development of B Cells.
The B-cell receptor complex is made up of cell-surface immunoglobulin with one each of the invariant signaling proteins Igα and Igβ. The immunoglobulin recognizes and binds an antigen but cannot itself generate a signal. It is associated with antigen-nonspecific signaling molecules—Igα and Igβ. Each has a single ITAM (immunoreceptor tyrosine-based activation motif) in its cytosolic tail that enables it to signal when the B-cell receptor is ligated with antigen. Igα and Igβ form a disulfide-linked heterodimer that is non-covalently associated with the heavy chains.
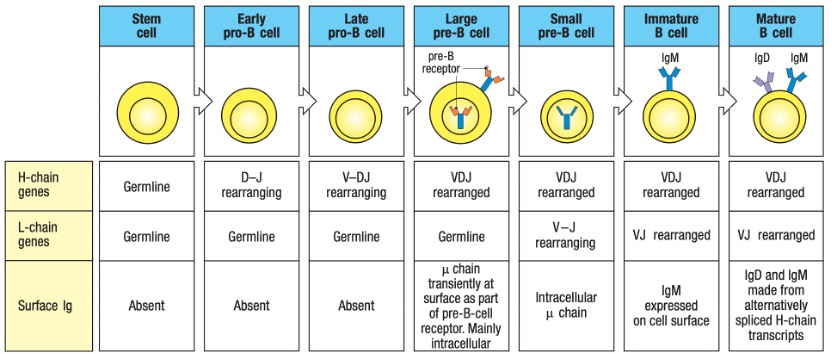
Figure 2. Heavy Chain (H Chain) Locus Rearrangements in the Development of B Cells.
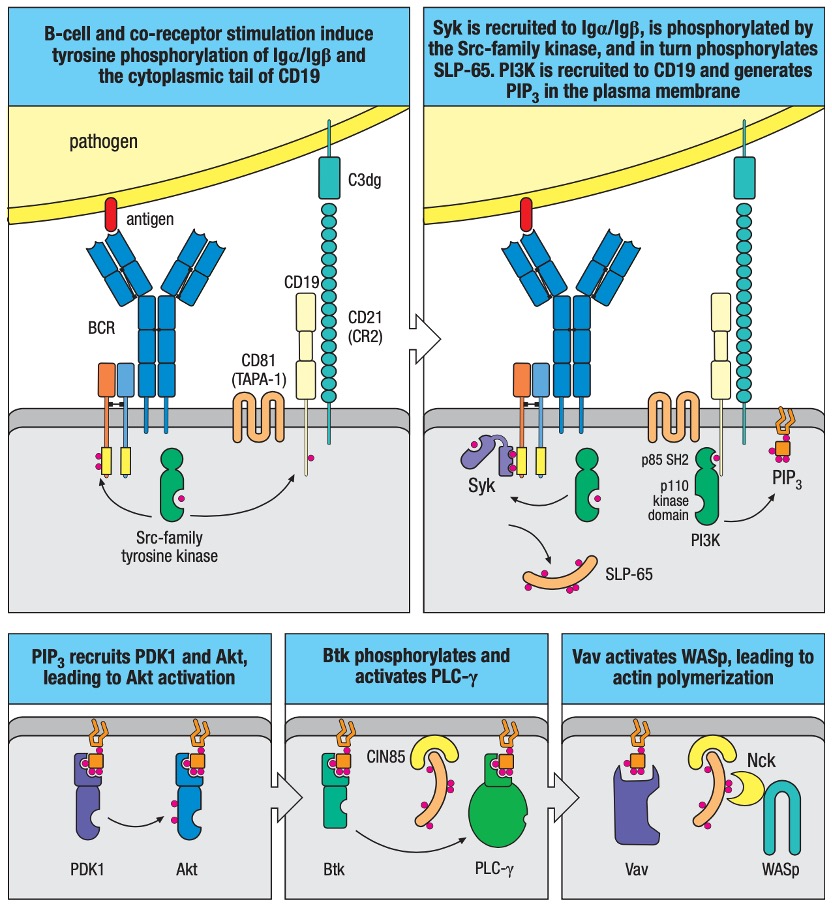
Figure 3. B-cell Signaling after Activation by Antigen in Peripheral Lymphoid Organs.
B-cell antigen receptor plus co-receptor engagement activates downstream signaling modules leading to activation of Akt, PLC-γ, and WASp. B-cell receptor (BCR) signaling is greatly enhanced when the antigen is tagged by complement fragments, engaging the B-cell co-receptor together with the B-cell antigen receptor. The B-cell co-receptor complex includes CD21 (complement receptor 2, CR2), CD19 and CD81 (TAPA-1). More details of the downstream B-cell signaling are displayed in Figure 3.
According to various isotypes or classes of antibodies, the suffix indicates the particular heavy chain present in the antibody: α (alpha), γ (gamma), δ (delta), ε (epsilon), μ (mu) heavy chain types correspond to IgA, IgG, IgD, IgE, IgM. (See Figure 4) A summary of the development of human B cells and the link between the different types of antibodies and different periods of B cells is demonstrated in Figure 5.
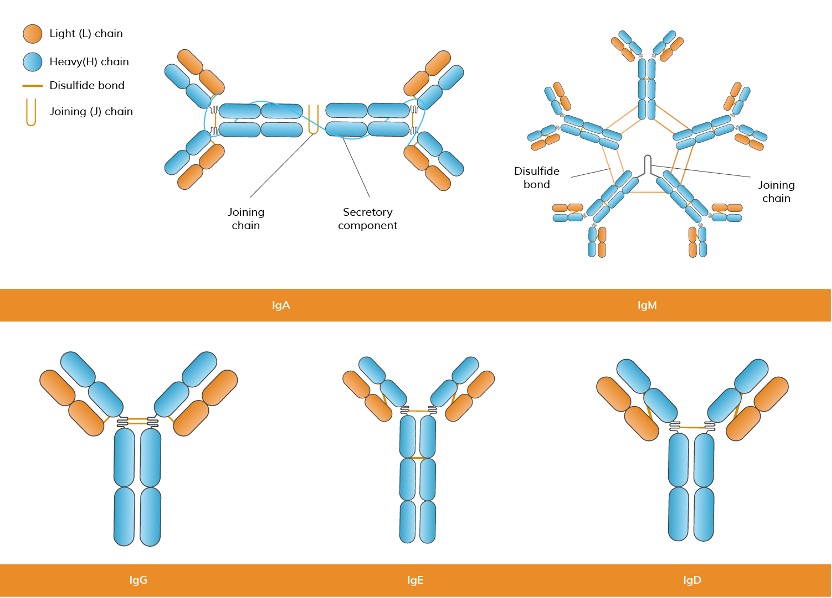
Figure 4. Classification of Antibodies Based on the Structures of Heavy Chains.
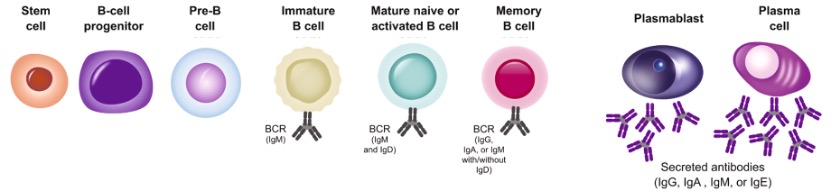
Figure 5. Different Kinds of Secreted Antibodies in Different Periods of the Development of B Cells
Reference
- Janeway. Immunobiology. Ninth Edition, CHAPTER 7 & CHAPTER 8.
- Treanor, B. B-cell receptor: from resting state to activate. Immunology. 2012, 136(1), 21-7.
T-cell signaling pathways
JIANG Hao, 4 Sept 2025
The ability of the T cell to respond to foreign pathogens appropriately depends heavily on signaling by cell-surface receptors, whose abnormal behavior would cause many diseases including immunodeficiency diseases and autoimmune diseases.
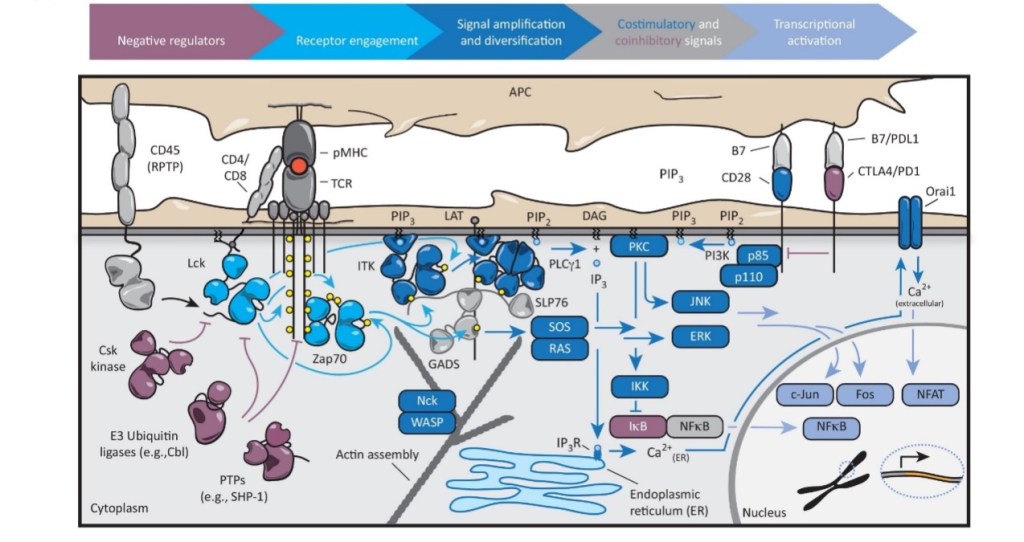
Figure 1. Overview of the T-cell signaling pathways.
The whole network of signaling pathways is highly complicated, efficiently organized and strictly regulated. Although many various pathways are involved, they share some common features and strategies, including the generation of second messengers such as calcium and phosphoinositide, the activation of both serine/threonine and tyrosine kinases, recruitment of signaling proteins to the plasma membrane and the assembly of multiprotein signaling complexes.
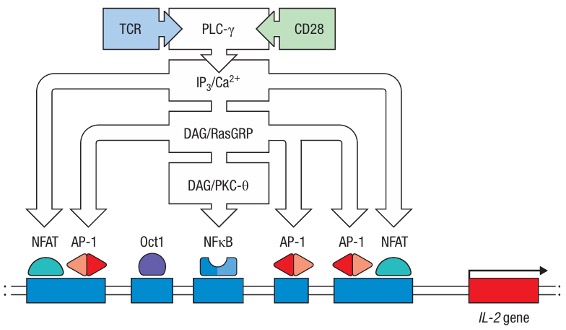
Figure 2. Simplified scheme about multiple signaling pathways necessary for IL-2 expression.
Profound impact would be cast by the signal transduction, such as the activation of transcription factors that lead directly or indirectly to the proliferation, differentiation, and effector function of activated lymphocytes. Take IL-2 expression as a typical example. TCR-CD28 two signal activation would lead to the binding of AP-1, NFAT, and NFκB to the promoter of the IL-2 gene through 3 separate pathways, so that the production of the cytokine IL-2 is started. Cell functions such as migration and shape changes are also mediated by changes in the cytoskeleton caused by signal transduction.
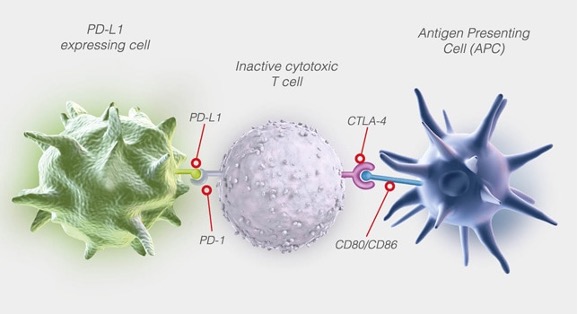
Figure 3. T-cell could be inhibited by PD-L1 or CTLA-4 pathway
The signaling pathways are also strictly regulated by some inhibitory receptors such as CTLA-4 and PD-1, which are crucial for the orderly functioning of the immune system, but also results in exhaustion in some pathological cases.
It’s notable that we are only beginning to understand the basics of these extremely complicated and wonderfully evolved signaling pathways, and this essay is even not enough to be called a brief introduction. The future would be exciting if all the mysteries could be fully unveiled and more effective therapies could be developed with the comprehensive understanding of the immune signaling pathways.
Reference
- Courtney, A. H., Lo, W. L., & Weiss, A. (2018). Trends in biochemical sciences, 43(2), 108-123.
- Janeways_Immunobiology_9th_Edition CHAPTER 7
Introduction to T cell receptor and major histocompatibility complex (MHC)
XU Zhiyi, 2 Sept 2025
The α:β T-cell receptor, which is the receptor for antigen recognition on most T cells, is composed of two protein chains, TCRα and TCRβ, and resembles in many respects a single Fab fragment of immunoglobulin. TCR could not survive without CD3γ, CD3δ, CD3ε, and ζ chains. Interactions are mediated by reciprocal charge interactions between basic and acidic intramembrane amino acids of the receptor subunits.
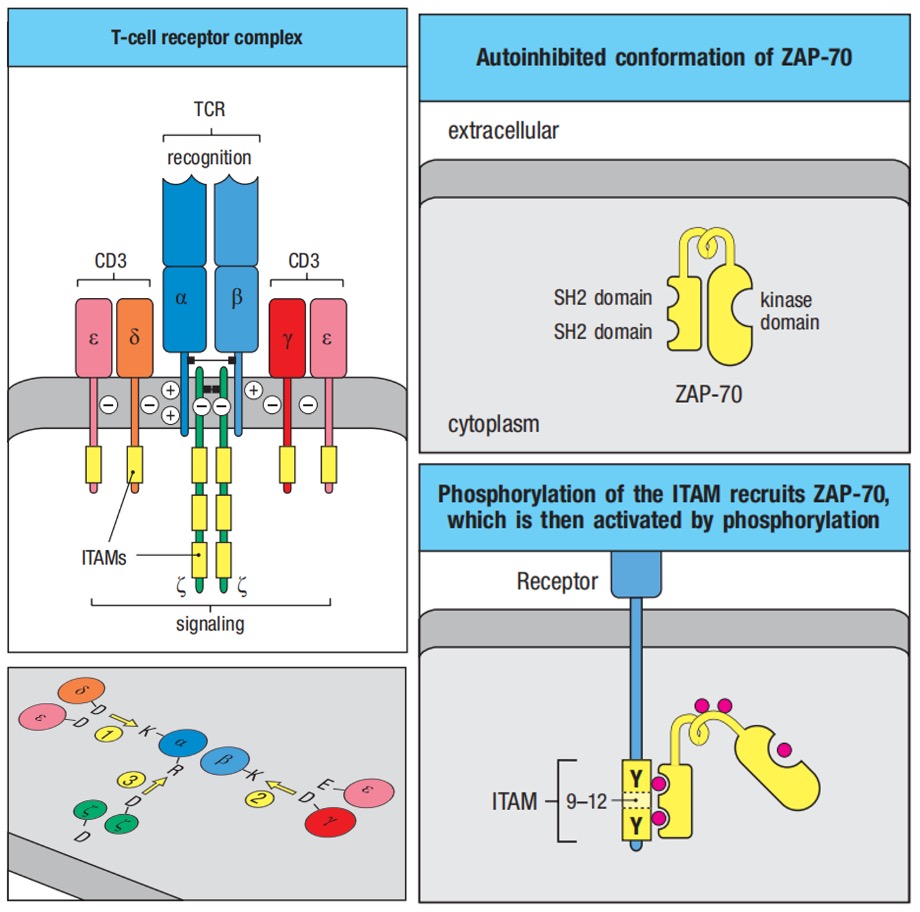
Figure 1. T cell receptor complex
α:β T-cell receptors are always membrane-bound and recognize a composite ligand of a peptide antigen bound to an major incompatibility complex (MHC) molecule. There are two classes of MHC molecules, MHC class I molecules presented on almost all the cells and MHC class II molecules presented only on the antigen presenting cells. Each MHC molecule binds a wide variety of different peptides. The peptide antigen is generated intracellularly, and is bound stably in a peptide-binding cleft on the surface of the MHC molecule.
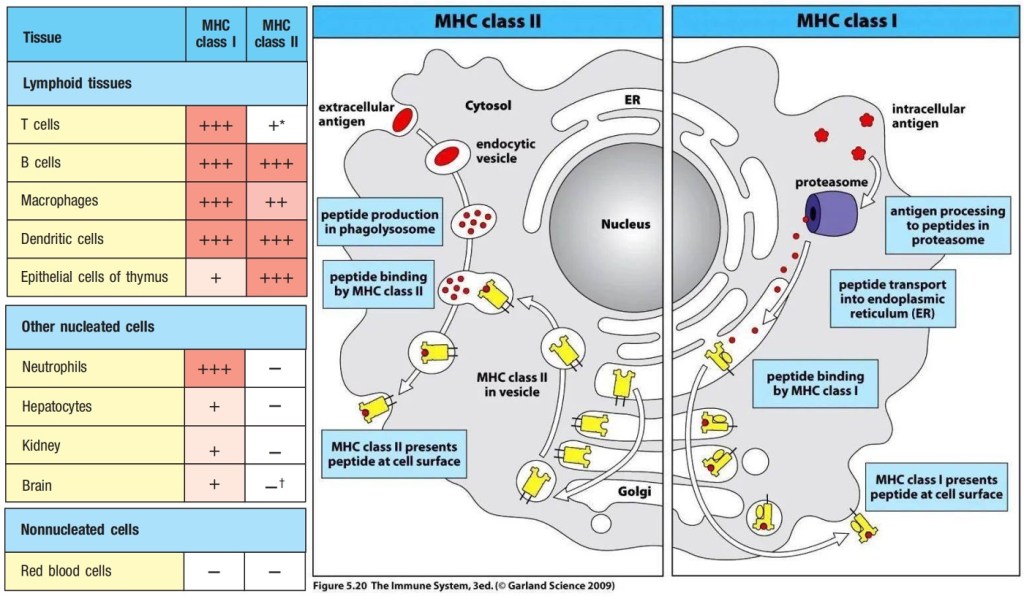
Figure 2. Major Incompatibility Complex (MHC) class I and class II
The MHC molecules are bound in their nonpolymorphic domains by CD8 and CD4 molecules that distinguish two different functional classes of α:β T cells. CD8 binds MHC class I molecules and can bind simultaneously to the same peptide:MHC class I complex being recognized by a T-cell receptor, thus acting as a co-receptor and enhancing the T-cell response; CD4 binds MHC class II molecules and acts as a co-receptor for T-cell receptors that recognize peptide:MHC class II ligands. A T-cell receptor interacts directly both with the antigenic peptide and with polymorphic features of the MHC molecule that displays it, and this dual specificity underlies the MHC restriction of T-cell responses.
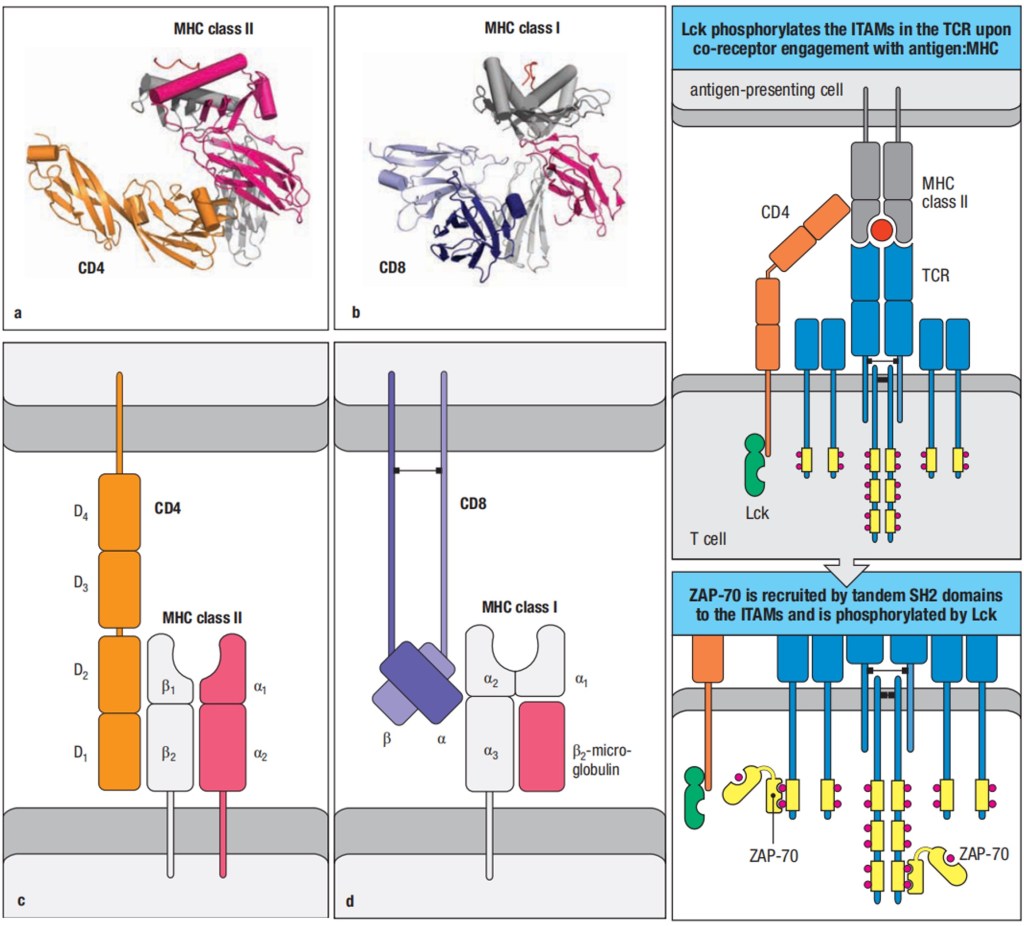
Figure 3. The Cooperation of CD4 and CD8 with peptide:MHC:T-cell receptor complexes in T cell activation
Reference
- Punt, J.; Stranford, S.; Jones, P. P.; Owen, J. A. Kuby Immunology, 8th ed.; Macmillan Education: New York, 2019.
- Murphy, K. M.; Weaver, C.; Mowat, A.; Berg, L.; Chaplin, D.; Janeway, C. A.; Travers, P.; Walport, M. Janeway’s Immunobiology, 9th ed.; New York London Gs, Garland Science, Taylor & Francis Group, 2016.
Protein Folding and diseases
Ryan Pak Hong Yip, 27 Aug 2024
Normally, misfolded proteins are either refolded or degraded as part of the cell’s quality control mechanisms. However, in cases where protein folding goes awry, it can lead to the development of diseases. Two notable examples are cystic fibrosis and prion diseases. Cystic fibrosis is a genetic disorder caused by mutations affecting the folding of the cystic fibrosis transmembrane conductance regulator protein (CFTR). Misfolded CFTR leads to the accumulation of thick, sticky mucus in the body’s airways and digestive system, particularly impacting the lungs. Prion diseases, such as Creutzfeldt-Jakob Disease (CJD), Kuru, mad cow disease, and scrapie, result from the misfolding of prion proteins. Unlike cystic fibrosis, there are currently no effective treatments or vaccines for prion diseases. These conditions can lead to spongiform encephalopathy, characterized by brain shrinkage, dementia, loss of appetite, and aggressive behaviors. Unfortunately, patients affected by prion diseases typically face a fatal outcome.

Figure 1. The CDJ disease.
Prion diseases exhibit symptoms akin to other neurodegenerative conditions but stand out as contagious illnesses that progress rapidly and often culminate in death. The misfolded prion protein PrP serves as an infectious agent, acting as a chaperone capable of propagating by binding to normal PrP and inducing its transformation into a hazardous conformation mirroring its own. The accumulation of these misfolded proteins leads to the onset of spongiform encephalopathy.
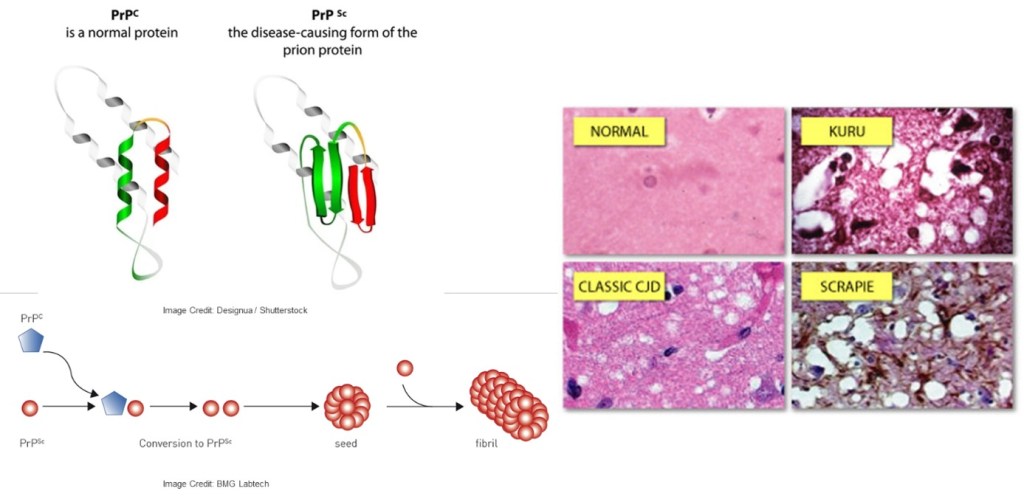
Figure 2. Misfolding of prion leads to neuronal degeneration.
One of the most infamous prion disease outbreaks occurred in the UK during the 1990s. The initial human fatality from mad cow disease was linked to the consumption of contaminated beef products, subsequently identified as variant CJD. The rapid spread of BSE among cattle and its entry into the food chain was facilitated by the use of feed containing scrapie-infected sheep meat. Prions exhibit an extraordinary level of infectivity, impervious to heat treatment, and prion diseases typically result in near-inevitable fatality. Preventing the transmission of prion diseases necessitates conducting BSE tests, culling infected animals, and imposing bans on materials posing a risk of transmitting BSE through animal and human food supplies, as well as other products.

Figure 3. The spreading of mad cow disease.
Reference
- Nickson AA, Clarke J. What lessons can be learned from studying the folding of homologous proteins? Methods. 2010 Sep;52(1):38-50. doi: 10.1016/j.ymeth.2010.06.003. Epub 2010 Jun 4. PMID: 20570731; PMCID: PMC2965948.
- Englander SW, Mayne L. The nature of protein folding pathways. Proc Natl Acad Sci U S A. 2014 Nov 11;111(45):15873-80. doi: 10.1073/pnas.1411798111. Epub 2014 Oct 17. PMID: 25326421; PMCID: PMC4234557.
- Hayer-Hartl M, Bracher A, Hartl FU. The GroEL-GroES Chaperonin Machine: A Nano-Cage for Protein Folding. Trends Biochem Sci. 2016 Jan;41(1):62-76. doi: 10.1016/j.tibs.2015.07.009. Epub 2015 Sep 25. PMID: 26422689.
- Han W, Christen P. Mechanism of the targeting action of DnaJ in the DnaK molecular chaperone system. J Biol Chem. 2003 May 23;278(21):19038-43. doi: 10.1074/jbc.M300756200. Epub 2003 Mar 24. PMID: 12654915.
- Kovač V, Čurin Šerbec V. Prion Protein: The Molecule of Many Forms and Faces. Int J Mol Sci. 2022 Jan 22;23(3):1232. doi: 10.3390/ijms23031232. PMID: 35163156; PMCID: PMC8835406.
- Gill AC, Castle AR. The cellular and pathologic prion protein. Handb Clin Neurol. 2018;153:21-44. doi: 10.1016/B978-0-444-63945-5.00002-7. PMID: 29887138.
- Creutzfeldt-Jakob disease: Rare cause of dementia-Creutzfeldt-Jakob disease – Symptoms & causes – Mayo Clinic. 2023 January 28. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/creutzfeldt-jakob-disease/symptoms-causes/syc-20371226#symptoms
Introduction to RNA-editing
XU Zhiyi, 13 Aug 2024
RNA Editing Therapy is a Rising Star in Medicine
RNA editing technology has evolved significantly over the past few decades. Initially, the focus was on understanding the basic mechanisms of RNA editing, particularly the adenosine-to-inosine (A-to-I) and cytidine-to-uridine (C-to-U) conversions. These discoveries laid the groundwork for developing therapeutic applications. Recently, RNA editing has gained momentum, with several therapies entering clinical trials. The COVID-19 pandemic highlighted the potential of RNA-based technologies, further accelerating research and development in this field. On June 18th, 2024, Roche payed Ascidian Therapeutics $42M to develop novel medicines for difficult to treat neurological diseases. The future of RNA editing looks promising, with ongoing advancements expected to enhance its precision, efficiency, and safety, potentially making it a cornerstone of personalized medicine.

Figure 1. Nature news of RNA-editing therapy
Mechanism of RNA Editing
RNA editing involves post-transcriptional modifications that alter the nucleotide sequence of RNA molecules. The most common types of RNA editing are A-to-I and C-to-U conversions. These processes are catalyzed by specific enzymes: adenosine deaminases acting on RNA (ADARs) for A-to-I editing and apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) for C-to-U editing. ADARs deaminate adenosine to inosine, which could pair with Cytidine and read as guanosine during translation, thereby altering the protein sequence. This A-to-I editing helps human body to distinguish between viral RNA and the host’s own RNA. ADAR1 could edit endogenous double-stranded RNA (dsRNA) by converting adenosine (A) to inosine (I), which is then recognized as self-RNA by the immune system. Compared with A-to-I editing, C-to-U editing is a less common but significant type of RNA editing in the human body. This kind of editing could change a codon or even create a premature stop codon during the translation to produce different proteins with one genes for different functions. One of the best-characterized examples of C-to-U RNA editing occurs in the mRNA of apolipoprotein B (ApoB). In the small intestine, a cytidine is edited to uridine (U), converting a CAA codon to a UAA stop codon. This results in the production of a shorter protein, ApoB48, which is essential for the assembly and secretion of chylomicrons. These mechanisms allow for the correction of genetic mutations at the RNA level without altering the underlying DNA sequence.

Figure 2. Mechanism of A-to-I and C-to-U RNA editing.
Advantages of RNA Editing Therapy Compared to CRISPR
RNA editing offers several advantages over DNA-based gene editing technologies like CRISPR-Cas9. One of the primary benefits is its transient nature. RNA molecules are naturally degraded and replaced, reducing the risk of permanent off-target effects. This makes RNA editing potentially safer, as any unintended modifications are not perpetuated in the genome. Additionally, RNA editing does not involve creating double-strand breaks in DNA, which can lead to unintended mutations and genomic instability. This makes RNA editing a more flexible and less invasive approach for therapeutic interventions. Moreover, RNA editing does not need exogenous or foreign enzymes like CRISPR-Cas enzymes from bacteria, which may pose immunological risks or require delivery with dual AAV vectors.
Application Examples of RNA Editing Technology
RNA editing has shown potential in various therapeutic applications. For instance, single-base RNA editing is being explored to treat genetic disorders such as alpha-1 antitrypsin deficiency (AATD), which affects the lungs and liver. One ADAR-based SERPINA1/AAT mRNA editor drug was already in Phase I clinical trials, while another three more RNA-editing drugs for AATD was in the preclinical stage. Another promising application is in the field of oncology, where RNA editing can be used to modify the expression of oncogenes and tumor suppressor genes, offering a novel approach to cancer treatment. Additionally, RNA editing has potential applications in neurological disorders, where it can be used to correct mutations associated with diseases like epilepsy and amyotrophic lateral sclerosis (ALS).
By leveraging the unique advantages of RNA editing, researchers are developing innovative therapies that could revolutionize the treatment of genetic diseases, offering new hope for patients worldwide.
Reference
- Lenharo M. Move over, CRISPR: RNA-editing therapies pick up steam. Nature. 2024 Feb;626(8001):933-934. doi: 10.1038/d41586-024-00275-6.
- Mullard A. RNA-editing drugs advance into clinical trials. Nat Rev Drug Discov. 2024 May;23(5):323-326. doi: 10.1038/d41573-024-00070-y.
- Zhang, D., Zhu, L., Gao, Y. et al. RNA editing enzymes: structure, biological functions and applications. Cell Biosci 14, 34 (2024). doi: 10.1186/s13578-024-01216-6.
- Lamers MM, van den Hoogen BG, Haagmans BL. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol. 2019 Jul 25;10:1763. doi: 10.3389/fimmu.2019.01763.
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321-49. doi: 10.1146/annurev-biochem-060208-105251.
- Barka A, Berríos KN, Bailer P, Schutsky EK, Wang T, Kohli RM. The Base-Editing Enzyme APOBEC3A Catalyzes Cytosine Deamination in RNA with Low Proficiency and High Selectivity. ACS Chem Biol. 2022 Mar 18;17(3):629-636. doi: 10.1021/acschembio.1c00919.
- Li Q, Gloudemans MJ, Geisinger JM, Fan B, Aguet F, Sun T, Ramaswami G, Li YI, Ma JB, Pritchard JK, Montgomery SB, Li JB. RNA editing underlies genetic risk of common inflammatory diseases. Nature. 2022 Aug;608(7923):569-577. doi: 10.1038/s41586-022-05052-x.
- Booth BJ, Nourreddine S, Katrekar D, Savva Y, Bose D, Long TJ, Huss DJ, Mali P. RNA editing: Expanding the potential of RNA therapeutics. Mol Ther. 2023 Jun 7;31(6):1533-1549. doi: 10.1016/j.ymthe.2023.01.005.
Aging of phase-separated proteins: liquids, gels, or solids
HAN Yongxu, 12 July 2024
The cellular environment includes various membraneless compartments, it can form and dissolve in response to cellular signals. The composition of condensates is specific. a cell can trigger phase separation of specific proteins, certain clients partition into these condensates, but others are kept out. Many of these compartments appear with phase transition. They have material properties of liquids, gels, or solids. Material properties are linked to distinct functions and pathologies. Condensate assembly is tightly regulated in the cellular environment, and failure to control condensate properties, formation, and dissolution can lead to protein misfolding and aggregation, human genetics indicate a strong link between condensate-forming proteins and ageing-related diseases such as neurodegeneration and cancer.

Fig. 1 (a) Functions of condensates and (b) Aging may be associated with aberrant condensates. [1]
In brief, Monomers can assemble into liquid-like condensates by phase separation. Liquid-like droplets can subsequently harden into a gel-like or glass-like state. They can further convert into solid-like fibrils. The ageing process is accompanied by condensate shrinking, suggesting that the molecular components increasingly interact and jam.
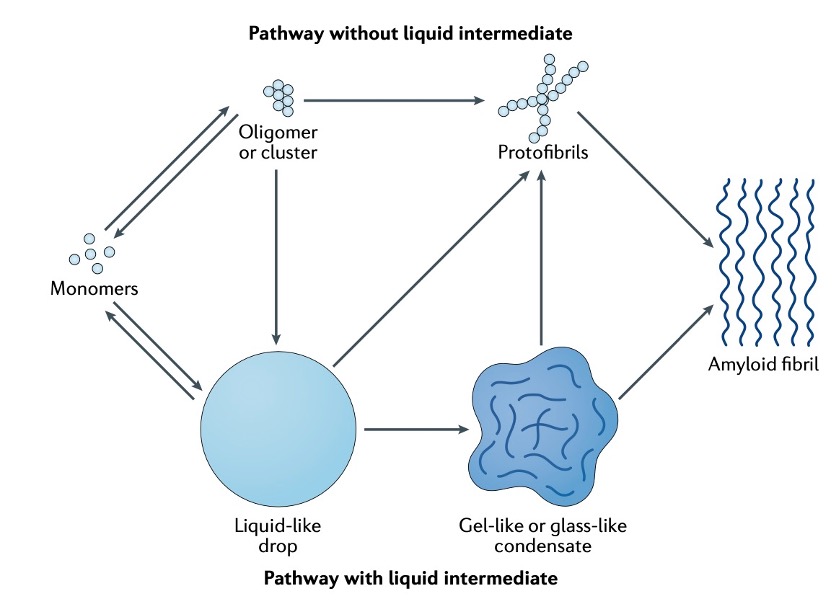
Fig. 2 Process of protein aging [1]
PTMs can modulate the phase transition. It can affect the affinity of the binding elements and dynamics of IDRs to regulate assembly and disassembly. Apart from the PTM, the aggregation of proteins like tau is highly dependent on its surrounding conditions. The negative charge of polyanions can compensate for the positive charge of tau, leading to an increase in aggregate. The metal ions can lead to oxidative stress with the generation of ROS and thus make protein aggregate. Fatty acids, as precursors of phospholipids in cellular membranes, can act as inducers of tau aggregation. The amphiphilic nature of fatty acids allows them to generate micelles. The anionic surfaces are enhanced by fatty acid micelles and could serve as nucleation points for tau fibrillization through charge neutralization effects.
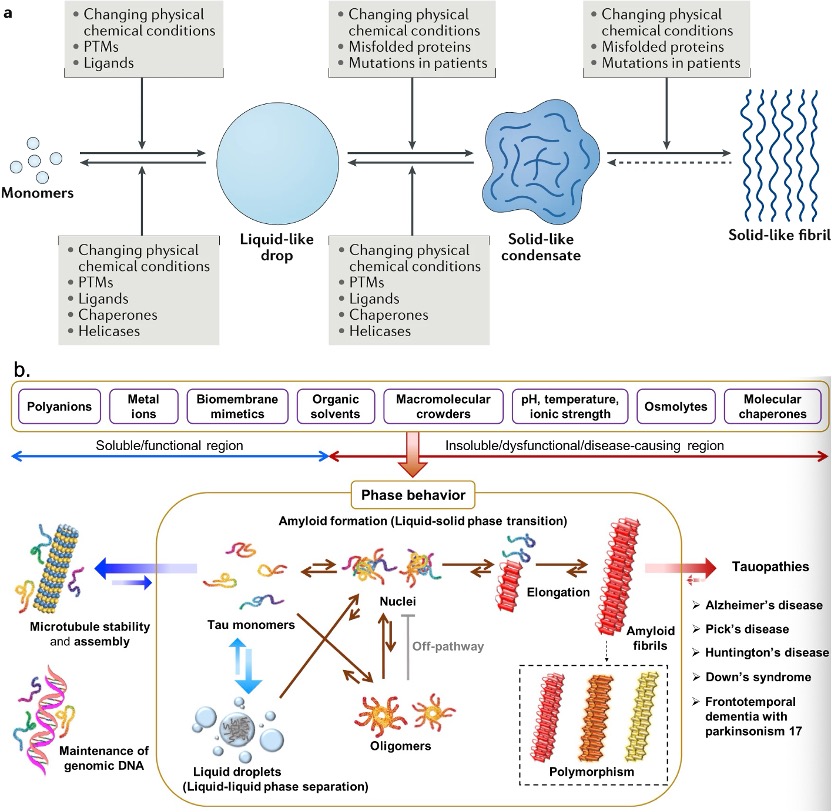
Fig. 3 The mechanisms and processes regulating the conversion of condensates. [2, 3]
Furthermore, pH, temperature, ionic strength, organic solvents, molecular crowders, and osmolytes can alter tau’s aggregation pathways and kinetics.
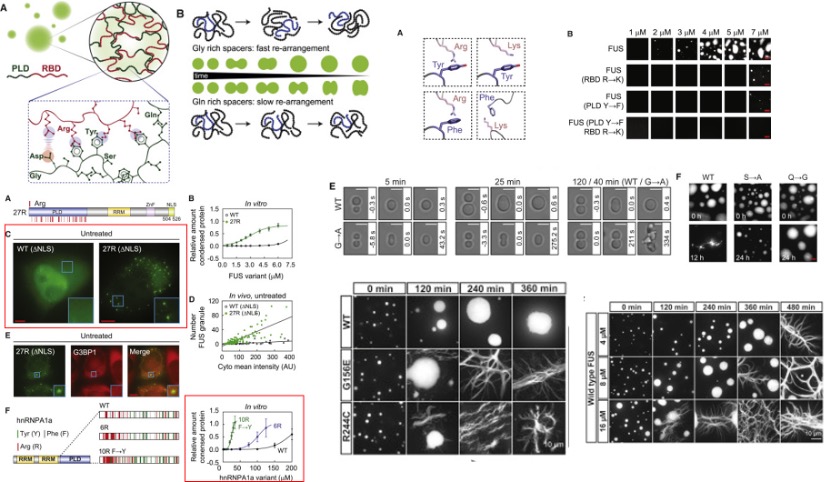
Fig. 4 The phase-separation behavior of FUS family proteins can be predicted by the prevalence and position of specific amino acids. [4]
Amino acids play an important role during phase transition. For hnRNPA1a, it normally phase separates at 100 μM, with the substitute of arginine and tyrosine, the saturation concentration was reduced to 15 μM. For FUS proteins, the result also shows the cation-π interactions from tyrosine-arginine interactions and the specific chemical structures of the tyrosine and the arginine side chains are all important in deciding the phase separation. Substitution of glycine for alanine in FUS (G→A) changes the fusion rate of two droplets. The (S→A) variant slowed hardening. But the (Q→G) variant produced droplets without change over 24 hr.
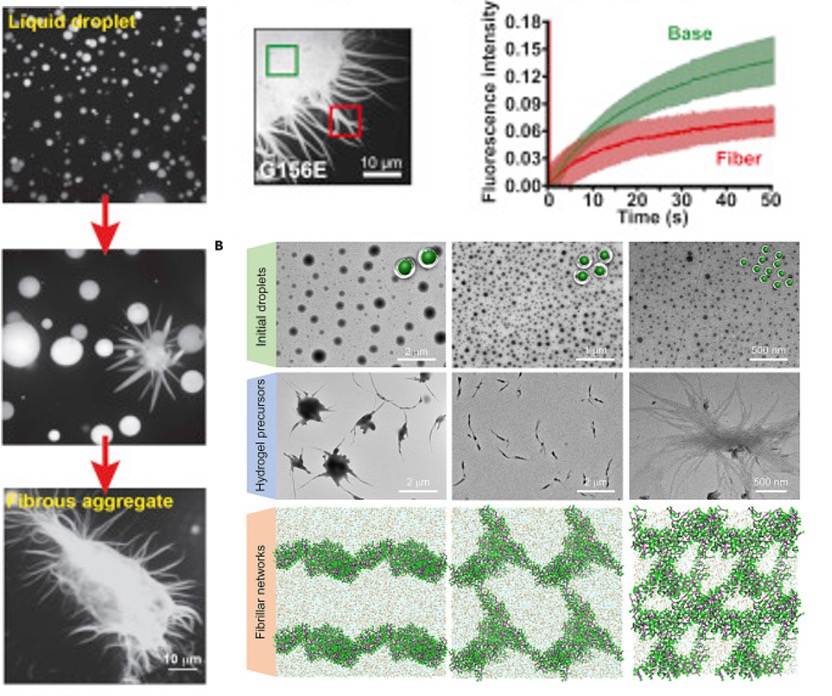
Fig. 5 The morphology of fibers. [5, 6]
For the morphology of the aggregate, they have short fibers and longer fibers. Small fibers like “sea urchins” and longer fibers like “starbursts.” The sea urchins showed that the fibers protruded from the surface and continuously grew until the starburst structure.
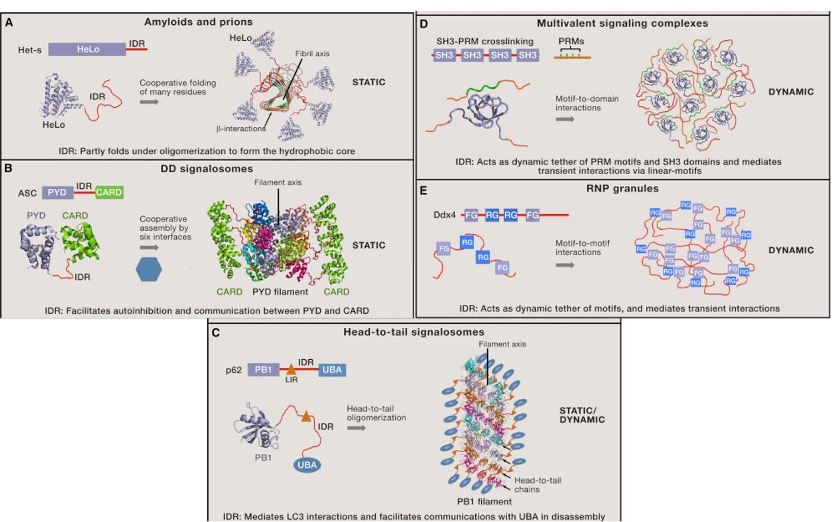
Fig. 6 Dynamic and static properties of condensate. [7]
The static and dynamic properties are associated with different material states. Generally, signaling events happen on short timescales, they require condensate that can rapidly form and dissociate. Since the dynamic properties favor the exchange of recruited proteins. But functional amyloids act as static scaffolds to either activate or inactivate the proteins. For prions, these structures are stable during cell division. In these cases, stability may be beneficial.
References:
- Alberti S, Hyman A A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nature Reviews Molecular Cell Biology, 2021, 22(3): 196-213.
- Nam G, Lin Y, Lim M H, et al. Key physicochemical and biological factors of the phase behavior of Tau. Chem, 2020, 6(11): 2924-2963.
- Murray D T, Kato M, Lin Y, et al. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell, 2017, 171(3): 615-627. e16.
- Wang J, Choi J M, Holehouse A S, et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell, 2018, 174(3): 688-699. e16.
- Patel A, Lee H O, Jawerth L, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell, 2015, 162(5): 1066-1077.
- Yuan C, Li Q, Xing R, et al. Peptide self-assembly through liquid-liquid phase separation. Chem, 2023, 9(9): 2425-2445.
- Wu H, Fuxreiter M. The structure and dynamics of higher-order assemblies: amyloids, signalosomes, and granules. Cell, 2016, 165(5): 1055-1066.
Recent Work about Targeted Protein Degradation
YUAN Dingdong, 5 July 2024
In 2001, Kathleen Sakamoto, Craig Crews and Ray Deshaies reported the first PROTAC, the PROTAC technology has been developed to degrade many kinds of proteins. The targeted protein degradation can be briefly divided into two categories by two degradation pathways: the first one is the traditional PROTAC small molecules which focus on the intracellular protein degradation using the proteasome pathway (Figure 1 left), and the other one is the cell-surface and secreted protein degradation using the lysosome pathway such as the LYTAC (Figure 1 right). [1 and 2] Many methods have been developed for different protein degradation using different strategies, but there are still many problems. For example, most methods had low selectivity to cancer cells (targeted cells), many proteins are hard to degrade, and the biosafety of the PROTACs is always judged. Therefore, people are still trying to develop novel methods to solve these problems. Here I hope to introduce three novel works on targeted protein degradation using different kinds of strategies.
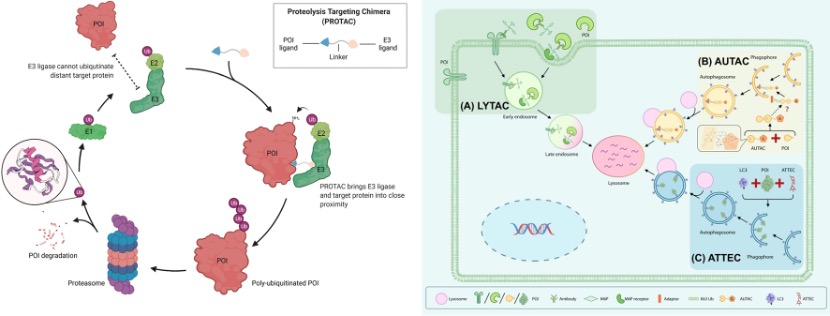
Figure 1. (left) PROTACs based on proteasome pathway. (right) PTD based on the lysosome pathway.
The first one is the TransTAC (Transferrin Receptor Targeting Chimeras). TransTAC is a bispecific antibody with one head that can bind to the targeted proteins and another head that can bind to the transferrin receptor 1(TfR 1). The TfR is required for iron import from transferrin into cells by endocytosis. Iron is an essential element for cells, and its transportation is facilitated by TfR 1. Rapidly dividing cells, such as cancer cells and activated immune cells, exhibit substantially increased TfR1 expression compared to non- or slow-dividing cells due to their high demand for iron. As a result, TfR1 is an attractive target for modulating these cell types. Several studies have investigated TfR1 as a therapeutic target for cancer with promising outcomes, as well as for tumor imaging and targeted drug delivery (Figure 2). [3]
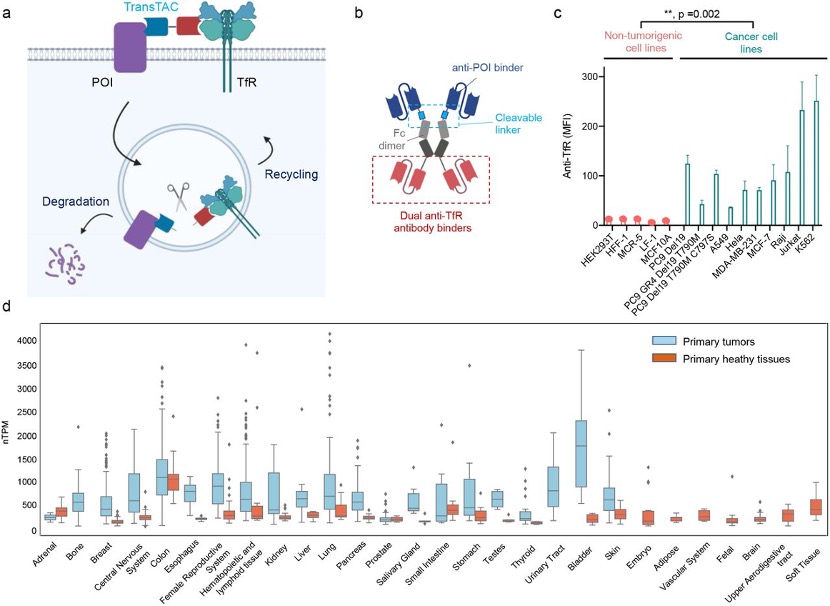
Figure 2. Overview of the TransTAC technology and Transferrin Receptor 1 (TfR1) expression analysis. (a) Schematic of TransTACs. TransTAC induces close proximity of TfR and a protein of interest (POI) at the cell surface. (b) Illustration of an example TransTAC protein. (c) Relative cell surface TfR1 expression levels across various non-tumorigenic and cancer cell lines characterized by flow cytometry. (d) Relative TFR1C RNA expression levels in primary tumor compared to normal tissues based on the MERAV database.
Then the authors designed and synthesized three bispecific antibodies for three cell surface proteins: PD-L1, ERFR, and CD20. From the western blot (WB) results, TransTACs all perform very well for the targeted protein degradation. Consequently, they developed TransTACs that may enable selective targeting of cancer cells while minimizing toxicity to normal cells, addressing the off-target effects commonly associated with traditional cancer treatments (Figure 3).
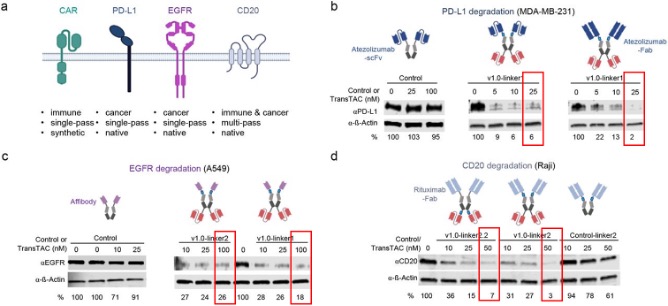
Figure 3. Developing TransTACs degraders for various proteins (a) Schematic of membrane proteins targeted by TransTACs in the present study. (b) PD-L1 degradation by TransTACs in MDA-MB-231 breast cancer cells analyzed by Western blot. A scFv or Fab format of atezolizumab is used as the PD-L1 binding moiety. (c) EGFR degradation by TransTAC in A549 lung carcinoma cells. An affibody is used as the EGFR binding moiety. (d) CD20 degradation by TransTAC. A Fab format of rituximab is used as the CD20 binding moiety.
The second one is the TransMoDEs. For most of the PROTACs, it is hard to cross the blood-brain barrier (BBB). But there are still many proteins that need to be degraded in the brain. TransMoDEs consist of two parts, the target binding ligand and the angiopep-2 peptide. The Angiopep-2 is a peptide derived from the Kunitz domain, which shows a high brain penetration capability to overcome the BBB. The angiopep-2 can help the TransTAC enter the brain cells and exocytosis the brain cells (Figure 4). [4]

Figure 4. Design of the TransMoDEs
Firstly, they modified the peptide with the biotin at different positions and used fluorescence-labeled streptavidin (SA) as the cargo. The flow cytometry results showed that the TransTAC with biotin at R3 position can help the SA enter into the two brain cell lines (Figure 5 left). Then, they also did the up-to-bottom and bottom-to-up experiments to mimic the TransTAC help the proteins cross the BBB. TransMoDEs represent the first molecules capable of inducing both the degradation of extracellular proteins and their bidirectional transcytosis across the BBB (Figure 5 right).

Figure 5. (left) TransMoDE facilitates uptake of streptavidin in mouse brain cell lines in a concentration and time-dependent manner. (right) Transcytosis of streptavidin (50 nM) across a BBB model using TransMoDE 3 (1000 nM).
The last one is the Supra-PROTAC. The Supra-PROTAC consist of three parts: the targeted protein binding ligand, E3 ligand, and a self-assembly peptide containing aryl sulfate esters. The peptide can be self-assembly to the nanoparticle which can enter the cells, the function of the PROTAC can be banned by the nanoparticle. However, when the nanoparticles enter the lysosome, the sulfatase can hydrolyze the sulfate esters, and the nanoparticles can be re-self-assembly to the nanofibers. The function of the degradation will recover (Figure 6). [5]

Figure 6. Schematic Illustration for In Situ Formulation of Supramolecular PROTACs (Supra-PROTACs) via Sulfatase-Induced Assembly of Peptides in Living Cells
The author showed two targeted proteins can be degraded by this method in cancer cells. Because of the lack of sulfatase in normal cells, the nanoparticles cannot be transferred to the nanofiber (Figure 7).
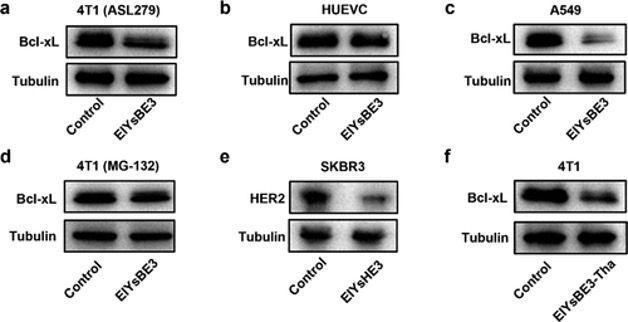
Figure 7. WB assays for the targeted protein degradation.
Although several ROS-triggered prodrugs and many drug delivery strategies have been developed, there are still problems that prevent the clinical application of these systems. Main challenges include biosafety of the materials, synthesis cost and biostability of the linkers. Besides, the hypoxia microenvironment of the tumour tissues may limit the efficiency of ROS production. Combination therapy has long been used to improve the effectiveness of cancer treatment. Besides, some compounds (called “ROS batteries”) that can store ROS in vitro and release ROS in vivo are being studied.
References
References
1. Ke Lia and Craig M. Crews. Chem. Soc. Rev., 2022, 51, 5214
2. Yu Ding, Yiyan Fei, and Boxun Lu. Trends Pharmacol. Sci., 2020, 41, 464
3. Dingpeng Zhang, Jhoely Duque-Jimenez, Garyk Brixi, Francesco Facchinetti, Kaitlin Rhee, William W. Feng, Pasi A. Jänne, and Xin Zhou. bioRxiv, 2023.08.10.552782.
4. Rebecca A. Howell, Shikun Wang, Mihir Khambete, David M. McDonald, and David A. Spiegel. J. Am. Chem. Soc., 2024, 146, 24, 16404–16411.
5. Ninglin Chen, Zeyu Zhang, Xin Liu, Hongbo Wang, Ruo-Chen Guo, Hao Wang, Binbin Hu, Yang Shi, Peng Zhang, Zhonghua Liu, and Zhilin Yu. J. Am. Chem. Soc., 2024, 146, 15, 10753–10766
Reactive oxygen species-triggered prodrug delivery strategies for cancer chemotherapy
KONG Hao, 24 June 2024
Cancer is still one of the deadliest diseases that have not been conquered. During the long history of fighting against cancers, several therapeutic methods have been developed, including surgery, radiation therapy, chemotherapy, immunotherapy and so on (Figure 1). Among these options, chemotherapy that uses highly cytotoxic small-molecule drugs remains a dominant method. By systemic administration of drugs, the proliferation of tumor cells can be inhibited, especially those in advanced or metastatic stages. Chemotherapy can be curative for certain types of cancers, especially in early stages. Besides, chemotherapy can be combined with other treatment methods like radiation therapy or targeted therapy to enhance effectiveness. Despite the great progress that has been made in small molecule drugs, they still suffer from challenges mainly associated with inherent small molecule characteristics, including low water solubility, poor targeting and short circulation in vivo.[1]
Figure 1. Options of cancer treatment methods that have been developed and the pros and cons of chemotherapy.
A series of strategies have been explored to overcome the drawbacks of chemotherapy, for example, the construction of stimuli-responsive activable prodrugs. Prodrugs are derivatives of drug molecules. The activity of the drugs is temporarily blocked by modifying the active sites to lower toxicity in normal tissues and can be activated in targeted cells by intracellular or extracellular stimuli like ROS, pH, enzymes, light or ultrasound, depending on the chemical structure of the cleavable linker (Figure 2A).[2-3] Moreover, encapsulation of small molecule prodrugs within various organic or inorganic nanocarriers has recently emerged as a highly potential strategy, as nanocarriers effectively shield the small-molecule drugs, resulting in an improvement in their stability, solubility, bioavailability and biodistribution. Drugs release can be triggered by various stimuli that are consistent with the prodrug activation (Figure 2B).[4]
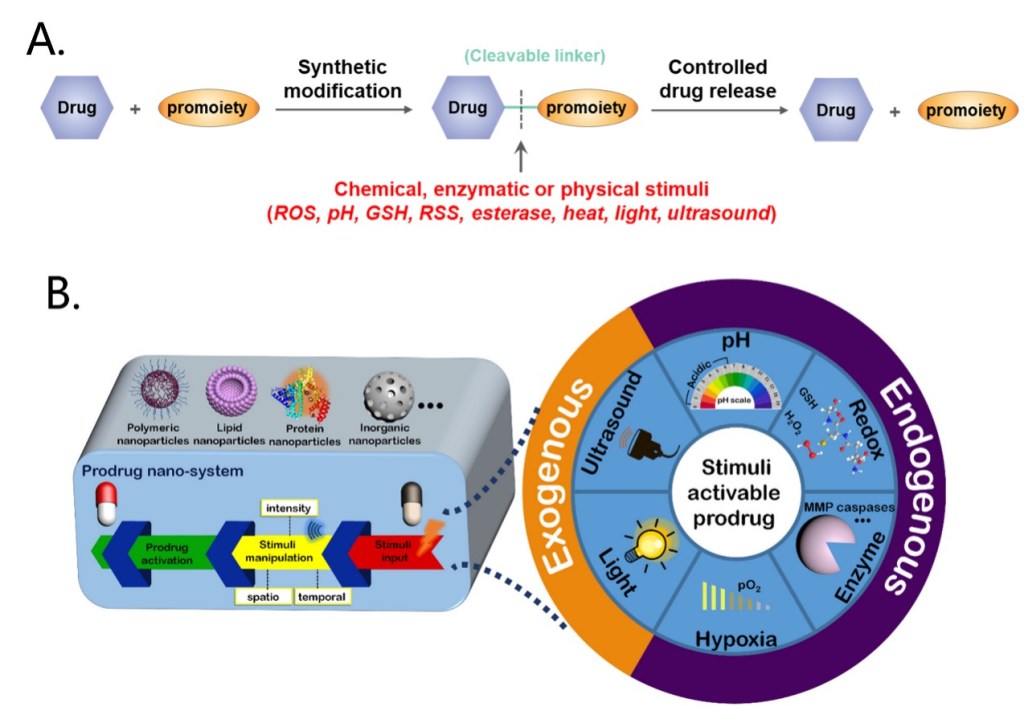
Figure 2. Overview of the prodrug construction strategies (A) and the nano-systems used for prodrug delivery (B).
Among these stimuli, reactive oxygen species (ROS) is a typical activation factor. ROS are transient oxygen intermediates produced by successive energy, electron or proton transfer of O2 (Figure 3A). Intracellular ROS are generated mainly in the mitochondria, peroxisome and endoplasmic reticulum, and in addition by some metabolic enzymes. Environmental factors such as radiation and xenobiotics can also cause ROS production (Figure 3B). ROS function as important signaling messengers in the regulation of cell functions, but excessive ROS production can cause cell damage and are associated with several diseases. For example, ROS levels in tumor cells are significantly higher than those in normal cells, which makes ROS as ideal markers for active targeting and inducing the controlled release of drugs.[5-7] Here we will list four common ROS-induced cleavage reactions and their applications in typical strategies for the construction of prodrug delivery systems.
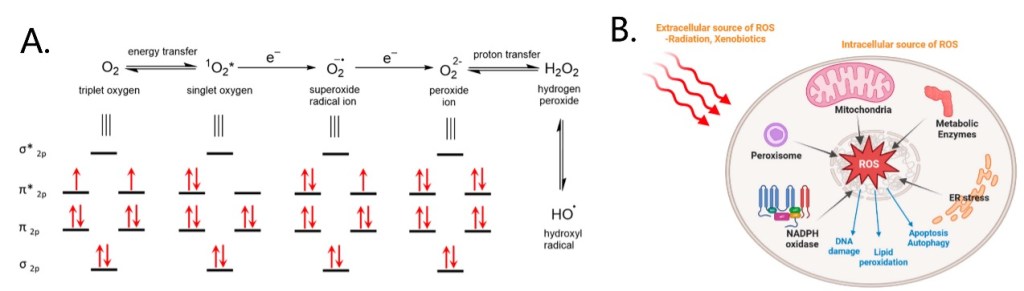
Figure 3. Production of ROS and molecular orbital of ROS (A) and sources and functions of intracellular ROS (B).
The first ROS-induced cleavage reactions used for prodrug construction is a 1O2 or H2O2 activated cleavage of thioketal linkers. Drugs can be totally released by subsequent reactions (Figure 4A). Xing’s group reported a cancer cell and mitochondria dual-targeting polyprodrug nanoreactor (DT-PN) that can achieve mtROS burst and induce cancer cell apoptosis. In this nanoreactor, Camptothecin (CPT) was selected as a model drug, which can act as a cellular respiration inhibitor to stimulate endogenous mtROS production and can promote cell apoptosis (Figure 4B).[8] The macromolecule PDMA-b-PCPTSM was covalently modified with a high content of repeating CPP units through the thioketal linker. Besides, cell-targeting cyclic RGD (cRGD) and mitochondria-targeting TPP were modified at the other side of the macromolecules. These modified macromolecules can co-assemble into nanoparticles, enter αvβ3 positive cancer cells and locate at mitochondria. Upon active targeting to mitochondria, endogenous upregulated mtROS in cancer cells can induce initial CPT release. In situ released CPT further triggers the circulating increase of mtROS, achieving subsequent amplification of high-dosage CPT release and a final mtROS burst, which are favorable for long-term high oxidative stress to efficiently eliminate cancer cells. Experimental results confirmed the CPP release from the nanoparticles, ROS burst in mitochondria both in vitro and in vivo. Also, these nanoparticles can target αvβ3 positive 4T1 cells and showed significant therapeutic effect on subcutaneous tumor in mice.
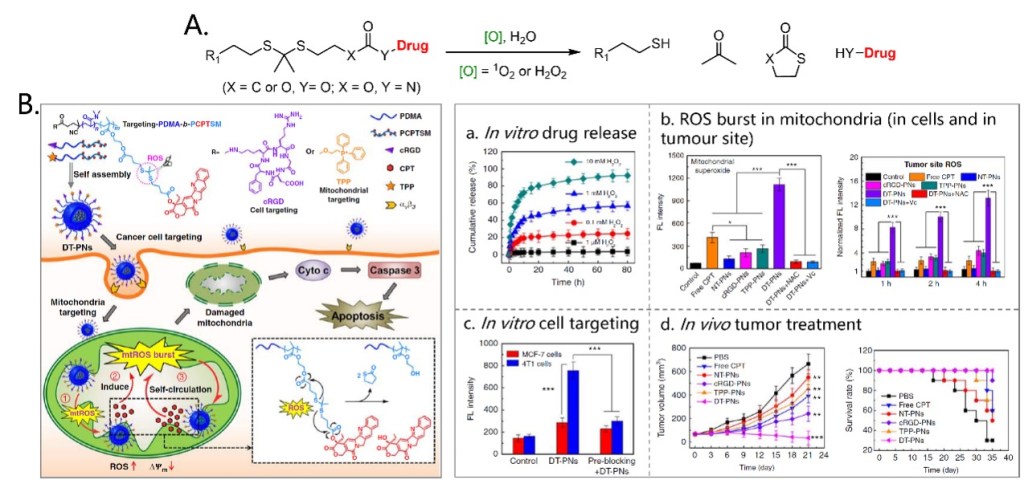
Figure 4. ROS-induced cleavage of thioketal linkers (A) and its application in a prodrug delivery nanoparticle system (B).
The second reaction is the 1O2 activated vinyldithioether cleavage reaction (Figure 5A). For example, Geng’s group reported a nanomedicine that combined chemotherapy, photodynamic therapy and pro-apoptosis to reduce the resistance to chemotherapy of pancreatic cancer cells (Figure 5B).[9] A pro-apoptotic peptide called Smac N7 was conjugated with a drug called Gem through a vinyldithioether linker. The mixture of the peptide-Gem molecules and the photosensitizer Ce6 can self-assemble into nanoparticles. These nanoparticles showed good performance on cellular uptake and 3D spheroids’ penetration and achieved synergistic therapeutic efficacy for pancreatic cancer with minimal systemic side effects.

Figure 5. ROS-induced cleavage of vinyldithioether linkers (A) and its application in a prodrug nanomedicine system (B).
The third reaction is the H2O2 induced phenylboronic acid oxidation. Drugs can be totally released by subsequent cascade reactions (Figure 6A). The work showed above employed light as the activation factor. But as we know light has limited penetration depth in vivo. Ultrasound is also a non-invasive physical stimulation, and it has much higher in vivo penetration depth than light. So, in a work reported by Huang’s group, ultrasound was used for activation of Ce6 to induce ROS production (Figure 6B).[10] A drug called SN38 was covalently modified using p-hydroxymethylboric acid to be block. The blocked drug called PBSN38 was loaded in a Ce6 modified liposome and used for murine colorectal cancer treatment. After ultrasound irradiation, SN38 can be released and can promote cancer cell apoptosis combined with ROS. On the murine colorectal cancer model, this system showed higher treatment efficiency and lower liver toxicity than a commercial drug CPT11.

Figure 6. ROS-induced cleavage of oxidation of phenylboronic acid (A) and its application in a prodrug delivery nano-liposome system (B).
The fourth reaction is a H2O2 induced peroxalate ester linker cleavage (Figure 7A). In a work reported by Ge’s group, CPT was conjugated with the backbone of a macromolecule through a peroxalate ester linker (Figure 7B).[11] Palmitoyl ascorbate (PA), which is one kind of the antioxidants, co-assembled with the CPT modified macromolecules to give micelles (named HPMs). This system can spontaneously promote hydrogen peroxide production by PA, then activate CPT release in the tumour microenvironment. No other chemical or physical stimuli are needed. Excess hydrogen peroxide and released CPT have been demonstrated to penetrate cancer cells efficiently, realizing synergistic in vitro and in vivo cancer cell killing.

Figure 7. ROS-induced cleavage of cleavage of a peroxalate ester linker (A) and its application in a prodrug delivery nano-micelle system (B).
Although several ROS-triggered prodrugs and many drug delivery strategies have been developed, there are still problems that prevent the clinical application of these systems. Main challenges include biosafety of the materials, synthesis cost and biostability of the linkers. Besides, the hypoxia microenvironment of the tumour tissues may limit the efficiency of ROS production. Combination therapy has long been used to improve the effectiveness of cancer treatment. Besides, some compounds (called “ROS batteries”) that can store ROS in vitro and release ROS in vivo are being studied.
References
[1] N. Behranvand, F. Nasri, R. Z. Emameh, P. Khani, A. Hosseini, J. Garssen and R. Falak. Cancer Immunol. Immun. 2022, 71, 507-526.
[2] X. Dong, R. K. Brahma, C. Fangc, S. Yao. Chem. Sci. 2022, 13, 4239-4269.
[3] J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman. Nat. Rev. Drug Discov. 2018, 17, 559-587.
[4] C. Ding, C. Chen, X. Zeng, H. Chen and Y. Zhao. ACS Nano 2022, 16, 13513-13553.
[5] P. T. Schumacker. Cancer cell 2015, 27, 156-157.
[6] Y. Hong, A. Boiti, D. Vallone and N. S. Foulkes. Antioxidants 2024, 13, 312.
[7] T. Chi, T. Sang, Y. Wang and Z. Ye. Bioconjugate Chem. 2024, 35, 1-21.
[8] W. Zhang, X. Hu, Q. Shen and D. Xing. Nat. Commun. 2019, 10, 1704.
[9] L. Zhu, S. Lin, W. Cui, Y. Xu, L. Wang, Z. Wang, S. Yuan, Y. Zhang, Y. Fan and J. Geng. Biomater. Sci. 2022, 10, 3624-3636.
[10] Y. Jiang, H. Chen, T. Lin1. C. Zhang, J. Shen, J. Chen, Y. Zhao, W. Xu, G. Wang and P. Huang. J. Nanobiotechnol. 2024, 22, 2.
[11] J. Li, W. Ke, L. Wang, M. Huang, W. Yin, P. Zhang, Q. Chen and Z. Ge. J. Control. Release 2016, 225, 64-74.
Phase Separation in Neurodegenerative Diseases
CHENG Kai 24 June 2024
Growing evidence has revealed that neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), Alzheimer’s disease (AD), and Parkinson’s disease (PD), are related to pathological protein aggregates. And abnormal liquid-liquid phase separation (LLPS) can cause protein aggregates. [1]
Figure 1. Four neurodegenerative diseases related to the different areas of the brain.
TDP-43 is one of the major pathological signature proteins in ALS disease[2]. Under normal physiological conditions, TDP-43 can participate in the formation of stress particles in response to changes in the cellular environment. Stress particles form membraneless structures through reversible LLPS, but mis-aggregation can lead to irreversible amyloid precipitation. The LLPS of TDP-43 mainly depends on the interaction of its carbon terminal helical structure domain. When this domain is destroyed and becomes unstable, normal LLPS cannot occur, protein aggregates are formed, then diseases further occur. Therefore, understanding the formation process and structural characteristics of these TDP-43 protein precipitates will provide new ideas for drug development in ALS diseases.
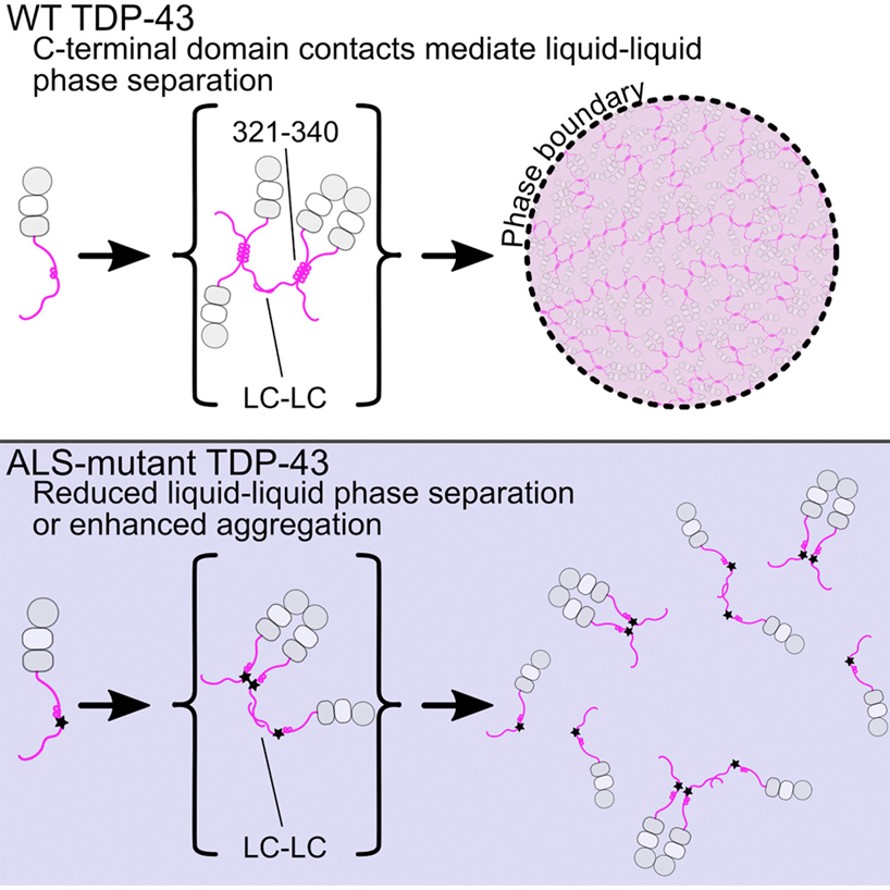
Figure 2 Liquid-liquid phase separation of TDP protein
The second, Fused in sarcoma (FUS) protein is an RNA-binding protein involved in RNA transcription, splicing, transport, and translation[3]. The occurance and progression of FTD disease are closely related to the abnormality of FUS. We know that FUS undergoes rapid, physiologically reversible phase separation between dispersed, liquid droplet, and hydrogel states. Research have showed that N-terminal low complexity domain of FUS is necessary and sufficient for phase separation and gelation of FUS, and does so by forming intermolecular β-sheet-rich fibrils. In this low complexity domain, cation-π interaction is considered to be a major phase separation driving force. Namely, carbon terminal arginine and nitrogen terminal tyrosine in FUS proteins can form phase separation through cation-π interaction. These dynamic phase structures take up, sequester, transport, and then release key RNA and protein cargos that regulate local RNA and protein metabolism in subcellular niches, such as axon terminals and dendrites. With the increase of cation-π interaction, the FUS tend to aggregate, the above processes may go awry, and disease will be triggered.
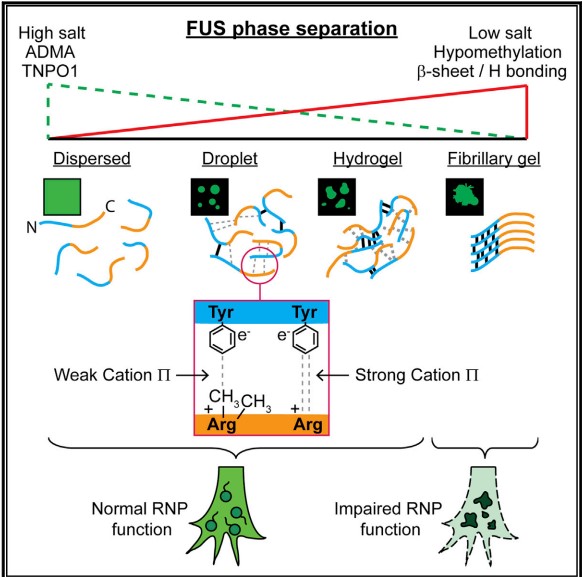
Figure 3 Liquid-liquid phase separation of the RNA-binding protein FUS
The third, Tau protein is the major constituent of neurofibrillary tangles in AD[4]. It have been proposed that LLPS of phosphorylated tau is a cell biologically relevant event, and tau droplet formation may resemble the initial step for aberrant tau aggregation in the brain. When phosphorylated and detached from the microtubules, the intraneuronal tau proteins can undergo LLPS to form droplets, and the transition from microtubule-bound and soluble monomeric/dimeric tau into tau droplets is facilitated by tau phosphorylation and the normal intracellular molecular crowding. When neurons are in a healthy state, LLPS may be a normal process of containing, condensing, and providing tau locally in the axon, and soluble and phase-separated tau exist in balance in the cytosol. When tau proteins are hyper-phosphorylated, equilibrium will shift in the direction of tau aggregation, these effects can cause high levels of short-term and long-term stress and eventually lead to neuronal death.
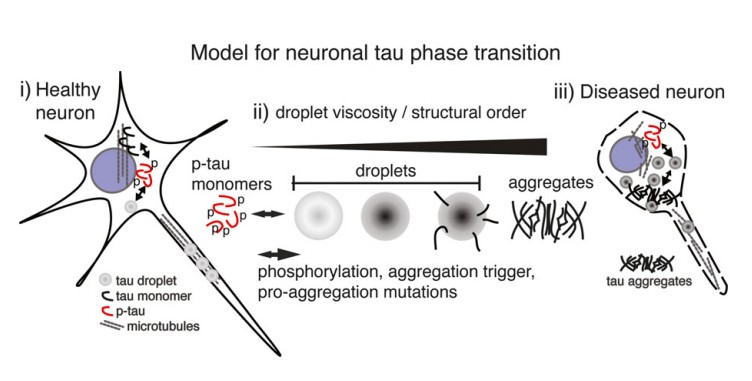
Figure 4 Liquid-liquid phase separation of Tau protein in neurons
The last, α-Synuclein is a natively unstructured protein, and its aggregation into cytotoxic oligomers and amyloid fibrils is associated with PD[5]. The α-Synuclein protein consists of three distinct domains: the N-terminal region, an aggregation-prone ‘non-amyloid-β component’ (NAC) and a flexible C-terminal domain, and the N terminal and hydrophobic NAC domain majorly drive α-Synuclein LLPS. Studies have showed that α-Synuclein form phase separation in early stage, and if some extreme conditions appeared, such as low pH, phospholation, etc., α-Synuclein droplets undergo a liquid to solid-like transition, which leads to a hydrogel formation that contains fibrillar aggregates, which is regulated by microtubules, and then PD will occur.
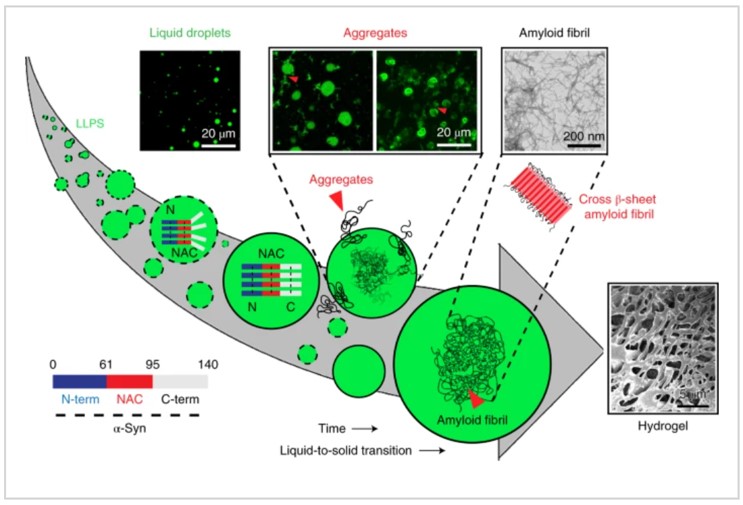
Figure 5. Liquid-liquid phase separation of α-Synucle protein
Although there are still many unsolved problems about LLPS in the field of neurodegenerative disease, this mechanism has undoubtedly revolutionized our understanding of neurodegenerative disease, and it may hold promise to be a new tool for treating diseases.
References
1. Wang. B., Zhang. L., Dai. T., et al. Liquid-liquid phase separation in human health and diseases. Signal Transduction and Targeted Therapy, 2021, 6(1): 290.
2. Conicella. A. E., Zerze. G. H., Mittal. J., et al. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure, 2016, 24(9): 1537-1549.
3. Qamar. S., Wang. G. Z., Randle. S. J., et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell, 2018, 173(3): 720-734. e15.
4. Wegmann. S., Eftekharzadeh. B., Tepper. K., et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. The EMBO journal, 2018, 37(7): e98049.
5. Ray. S., Singh. N., Kumar. R., et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nature chemistry, 2020, 12(8): 705-716.
Formation and Applications of Metal-Polyphenol Networks
XIAO Kemeng 18 June 2024
Polyphenols, the organic molecules containing phenolic hydroxyl groups, are ubiquitous in natural plants and have demonstrated beneficial effects on human health, such as anti-inflammatory, antimicrobial, and anti-cancer properties. The common polyphenols are summarized in Figure 1. Among these, tannic acid (TA) is of particular interest, as its adhesive capabilities enable it to readily deposit on a wide range of inorganic, organic, and biological substrates. The abundant di- and/or trihydroxy phenyl units in TA facilitate its easy surface modification. For instance, TA can serve as a hydrogen donor to form stronger hydrogen bonds with molecules containing hydrogen acceptors. Furthermore, the deprotonated phenolic groups in TA can establish robust associations with metal cations, an attribute that has garnered significant attention due to the attractive characteristics of these TA-metal complexes, such as structural flexibility, pH-responsiveness, and thermal stability. Notably, among various metal ions, Fe3+ ions, with their high binding affinity and low biotoxicity, have been a focus of particular interest in this regard. The pioneering work on TA-Fe assemblies, which successfully achieved versatile film and particle engineering, was first reported in 2013.[1] Building on this research, TA has since been coupled with various metals to enable self-assembly and subsequent coating of different materials. [2]
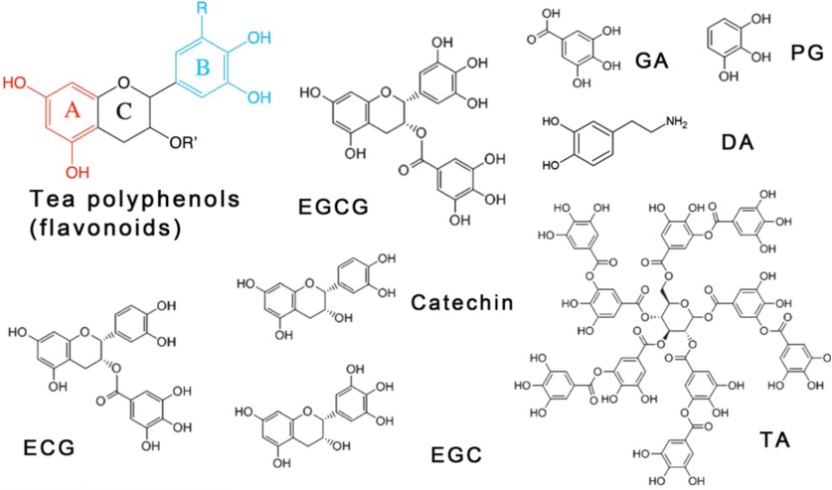
Figure 1. Chemical structures of common polyphenols.[3]
The TA-Fe coordination can be achieved through various methods (Figure 2). One approach involves the simple mixing of iron and polyphenols in the presence of a substrate, which is a straightforward, cost-effective, and scalable process. Alternatively, a multistep method can be employed, where polyphenols are first incubated with a template (e.g., polystyrene), followed by the addition of metal ions to the resulting polyphenol-terminated template. Repeating this two-step sequence leads to the deposition of multiple layers of metal-polyphenol networks (MPNs) on the template. MPNs prepared via the multistep method tend to have a higher metal ion proportion compared to the simple mixing approach. Additionally, the sol-gel method has also been utilized for MPN assembly. In this technique, the polyphenols are first pre-crosslinked by formaldehyde in alkaline water/ethanol solutions to fabricate phenolic oligomers, after which metal ions are added to the suspension of these polyphenol oligomers to form MPN nanospheres.
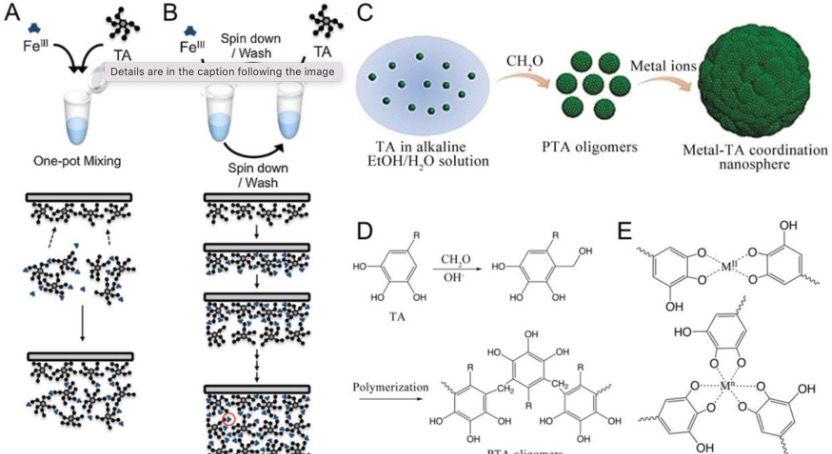
Figure 2. Different method for fabrication of MPN.[4]
The applications of Fe-TA networks are further explored, with several representative works highlighted. Firstly, Fe-TA networks have been utilized for the organized assembly of particles into superstructures (Figure 3). These superstructures are of great interest across a variety of fields due to their distinct mechanical, electronic, magnetic, and optical properties. This assembly method involves a two-step process: first, the particle surfaces are functionalized with polyphenol moieties, a step that has been shown to be largely independent of the substrate and/or particle surface chemistry, largely owing to the multidentate binding capabilities of polyphenols. This is followed by the directed assembly of the functionalized particles, driven by interfacial molecular interactions and interparticle locking mediated by metal ions. The right-hand figure demonstrates the successful assembly of a superstructure using small sphere-shaped building blocks.
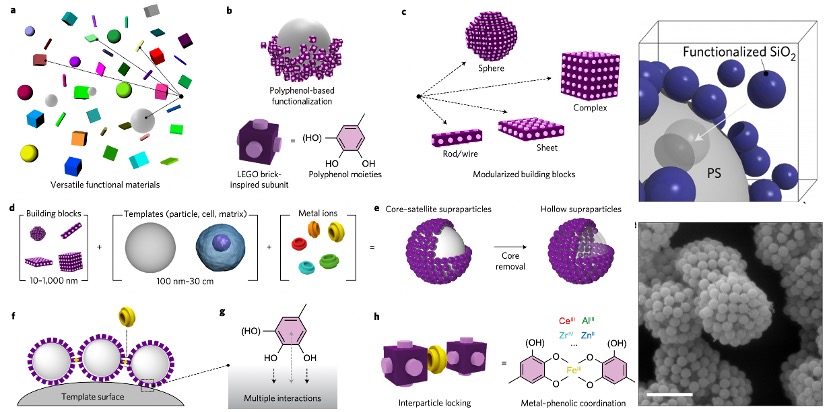
Figure 3. Schematic of versatile materials with different shapes, structures, compositions and functionalities to form superstructures via TA-Fe networks. Right: the images of formed superstructures on SiO2.[5]
A second application of Fe-TA networks is in the context of semi-artificial photosynthesis (Figure 4). Semi-artificial photosynthesis involves the integration of inorganic-biological hybrid systems, which offer the potential to serve as sustainable, efficient, and versatile chemical synthesis platforms. This approach combines the light-harvesting properties of semiconductors with the synthetic capabilities of biological cells. In one example, light-harvesting indium phosphide (InP) nanoparticles were independently prepared and coated with Fe-TA networks, which were then adhered to yeast cells. Owing to the close contact between the nanomaterials and the yeast cells, upon illumination, the nanomaterials could produce photoelectrons that were then transferred to the yeast cells. This, in turn, facilitated the regeneration of NADPH within the yeast cells, which was then harnessed to drive the biosynthesis of shikimic acid (SA).
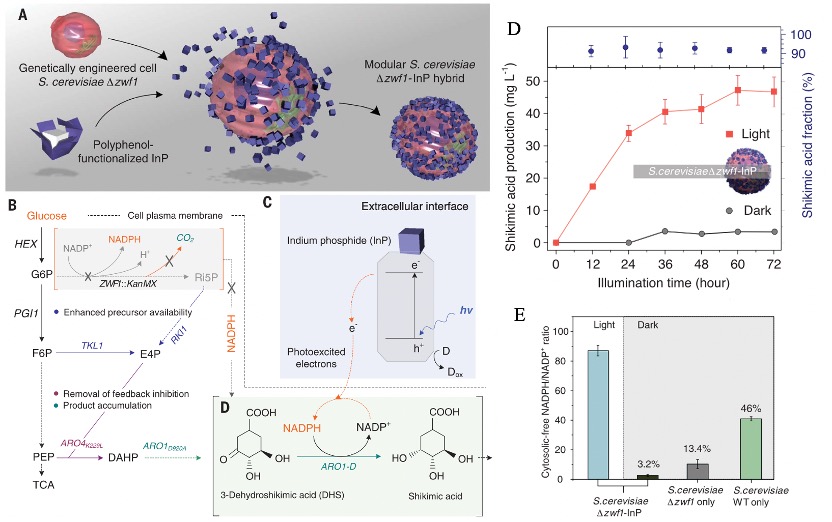
Figure 4. Assembly of S. cerevisiae-InP hybrids and rationally designed metabolic pathways for light-driven shikimic acid production.[6]
Furthermore, Fe-TA networks have also been utilized to protect bacterial cells (Figure 5). The physical encapsulation of beneficial bacteria within a protective shell presents an attractive approach to address the imprecise killing action of antibiotics on probiotics. In this work, probiotic cells were individually armored by a biocompatible supramolecular coating composed of TA and ferric ions. This safe and edible nanoarmor provided protection to both Gram-positive and Gram-negative bacteria against six clinically relevant antibiotics. The multiple interactions between the nanoarmor and antibiotic molecules allow the antibiotics to be effectively absorbed onto the nanoarmor, thereby prolonging the survival and colonization of the probiotic bacteria in the gut. This helps to restore a healthy balance in the gut microbiome.
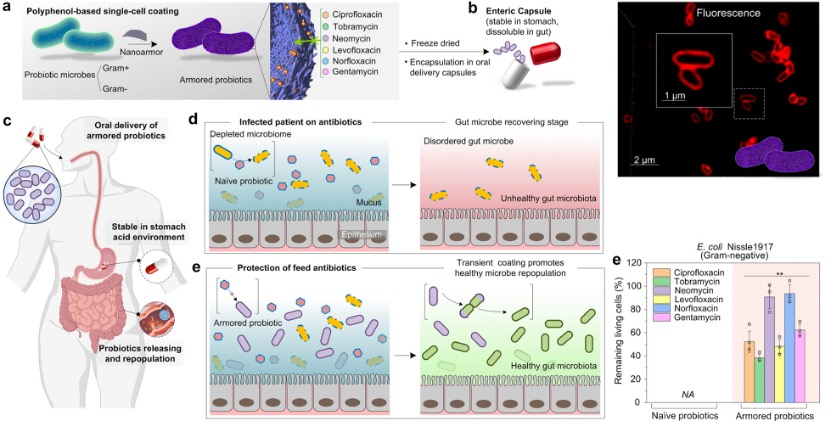
Figure 5. Natural polyphenol-based single-cell coating (nanoarmor) for the protection of bacteria from antibiotics in the gastrointestinal (GI) tract. [7]
Drawing a parallel to the protective effect observed on bacteria, a polyphenol-based cell surface functionalization strategy was constructed to enable the development of modular cancer vaccines (LMP vaccines) (Figure 6). In this approach, Mn2+ ions and TA were used to form a continuous nanocloak on cancer cells, which then enabled the decoration of immune agonist lipopolysaccharide (LPS) on the surface. This ensured the efficient delivery of the vaccine to antigen-presenting dendritic cells (DCs). Notably, after internalization by the DCs, the Mn2+ inherent to the nanocloak was able to sensitize the innate immune stimulator of the interferon gene (STING) pathway, promoting the generation of interferon-I (IFN-I). This facilitated the activation of both DCs and cytotoxic T cells. By harnessing the activation of both the innate and adaptive immune responses, the suppressive mechanism of the tumor microenvironment was effectively reprogrammed, which is beneficial for enhancing the recognition and killing of tumor cells.
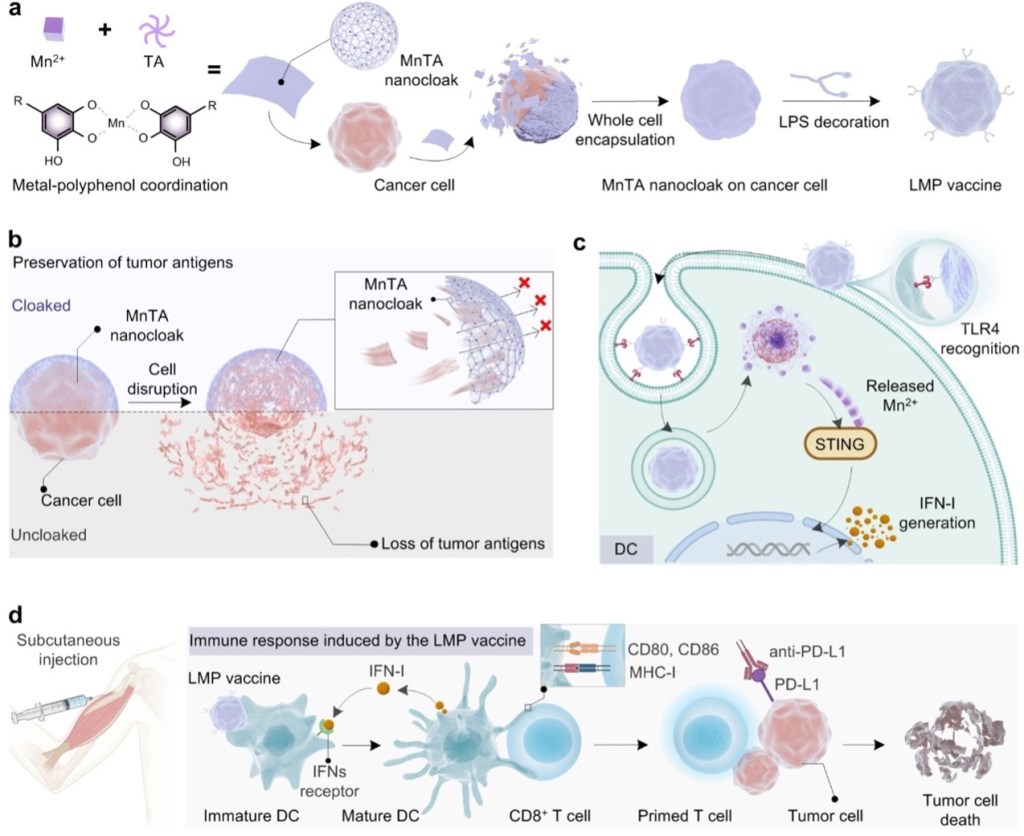
Figure 6. Schematic illustration of the LMP vaccine design and principle for STING pathway activation induced tumor inhibition in synergistic immunotherapy.[8]
Although various biomedical applications of TA-based metal-phenolic networks (MPNs) have been demonstrated and validated in laboratory settings, transitioning these materials from the lab to clinical use requires further investigation. Future researchers must delve into the detailed mechanisms and gain a deeper understanding of the specific functions of TA-based MPNs in biological applications. For instance, at the molecular level, the assembly mechanism underlying these materials remains poorly understood. Additionally, the structure-function relationship of the assemblies constructed with TA and multiple metal ions has not been thoroughly investigated. Elucidating these fundamental aspects is crucial to advancing the bio-applications of TA-based MPNs. Moreover, the modification of TA on substrates can directly influence the physicochemical properties and their associated functions. Therefore, it is of great significance to exert precise control over the thickness and self-assembly process of Fe-TA networks to ensure consistent performance and properties. In summary, future research should prioritize unraveling the molecular-level assembly mechanisms, establishing structure-function correlations, and optimizing the synthesis and assembly of TA-based MPNs. These efforts will be instrumental in bridging the gap between laboratory demonstrations and successful clinical translation of these promising biomaterials.
References
[1] H. Ejima, J.J. Richardson, K. Liang, J.P. Best, M.P. van Koeverden, G.K. Such, J. Cui, F. Caruso, One-step assembly of coordination complexes for versatile film and particle engineering, Science, 341 (2013) 154-157.
[2] J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J.J. Richardson, Y. Yan, K. Peter, D. Von Elverfeldt, C.E. Hagemeyer, Engineering multifunctional capsules through the assembly of metal–phenolic networks, Angewandte Chemie International Edition, 53 (2014) 5546-5551.
[3] Y. Guo, Q. Sun, F.-G. Wu, Y. Dai, X. Chen, Polyphenol-Containing Nanoparticles: Synthesis, Properties, and Therapeutic Delivery, Advanced Materials, 33 (2021) 2007356.
[4] J. Wei, G. Wang, F. Chen, M. Bai, Y. Liang, H. Wang, D. Zhao, Y. Zhao, Sol–gel synthesis of metal–phenolic coordination spheres and their derived carbon composites, Angewandte Chemie, 130 (2018) 9986-9991.
[5] J. Guo, B.L. Tardy, A.J. Christofferson, Y. Dai, J.J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R.R. Dagastine, Modular assembly of superstructures from polyphenol-functionalized building blocks, Nature nanotechnology, 11 (2016) 1105-1111.
[6] J. Guo, M. Suástegui, K.K. Sakimoto, V.M. Moody, G. Xiao, D.G. Nocera, N.S. Joshi, Light-driven fine chemical production in yeast biohybrids, Science, 362 (2018) 813-816.
[7] J. Pan, G. Gong, Q. Wang, J. Shang, Y. He, C. Catania, D. Birnbaum, Y. Li, Z. Jia, Y. Zhang, A single-cell nanocoating of probiotics for enhanced amelioration of antibiotic-associated diarrhea, Nature Communications, 13 (2022) 2117.
[8] X. He, G. Gong, M. Chen, H. Zhang, Y. Zhang, J.J. Richardson, W.Y. Chan, Y. He, J. Guo, Metal‐Phenolic Nanocloaks on Cancer Cells Potentiate STING Pathway Activation for Synergistic Cancer Immunotherapy, Angewandte Chemie, 136 (2024) e202314501.
Bioorthogonal Reactions for Prodrug Activation
BAO Yishu, 6 June 2024
Prodrugs are derivatives of drugs designed to inactivate the drug. The drug can be reactivated by chemical or physical stimuli. Chemical stimuli usually involve reactions between azide and phosphine, tetrazine and trans-cyclooctene (TCO), desilylation, and metal catalysts. Physical stimuli typically include NIR light, x-ray, gamma-ray, and ultrasound. Active drugs commonly used include doxorubicin, MMAE, and 5-FU.[1]
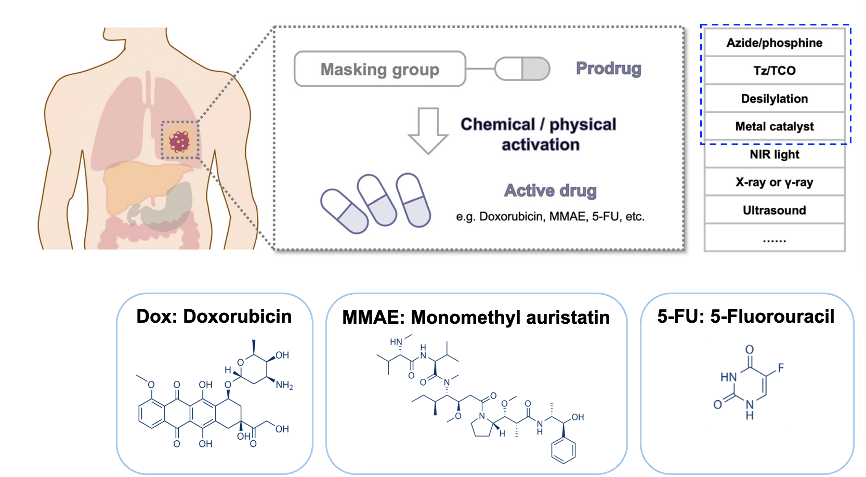
Figure 1. Overview of prodrug activation strategies for anticancer agents.
In 2006, Florent’s group reported the first biorthogonal cleavage reactions for prodrug activation.[2] They introduced the triarylphosphine motif to shield the amine group of doxorubicin. Azide serves as the trigger. The reaction was performed in a water and THF solution. Azide reacts with triarylphosphine, releasing N2, which then attacks the carbonyl carbon center, leading to a nucleophilic substitution reaction and the formation of a new C-N bond. Then, the oxygen minus facilitates the electron transfer. Over 90% of doxorubicin was released within 3 hours. TCO can also react with azide compounds.[3] It reacts with azides and initially undergoes 1,3-dipolar cycloaddition, forming an unstable 5-membered ring. Subsequently, the N-N bond breaks, resulting in electron transfer, release of N2, and formation of the C-N double bond. The resulting imine is unstable under acidic conditions, leading to hydrolysis and generation of the aldehyde and amine. The lone pair electrons on the N atom trigger 1,6-elimination, releasing the drug.
Although azide reductions are high biocompatibility, they exhibit relatively slow reaction rates. Therefore, increasing attention has been paid to the inverse electron-demand Diels–Alder reactions, also known as IEDDA reactions. The first IEDDA reaction was developed by the famous chemist Woodward in 1950 and was widely used in total synthesis. In 1959, Lindsey’s group reported the first IEDDA reaction between tetrazine and alkene. In 2013, Robillard’s group reported the first bioorthogonal IEDDA reaction.[4] They protected the Dox amine group using TCO, and tetrazine was used to trigger the reaction, releasing Dox and generating N2 and CO2 as by-products. This process also can be controlled by a photolysis reaction.[5] Photo-responsive groups can be introduced to the tetrazine ring. Only after tetrazine is decaged by 450nm light can it react with TCO. Results show that this light-active tetrazine prodrug therapy has similar cytotoxicity as dox itself, indicating that both the photolysis reaction and IEDDA reaction between the tetrazine and TCO are in high efficiency.
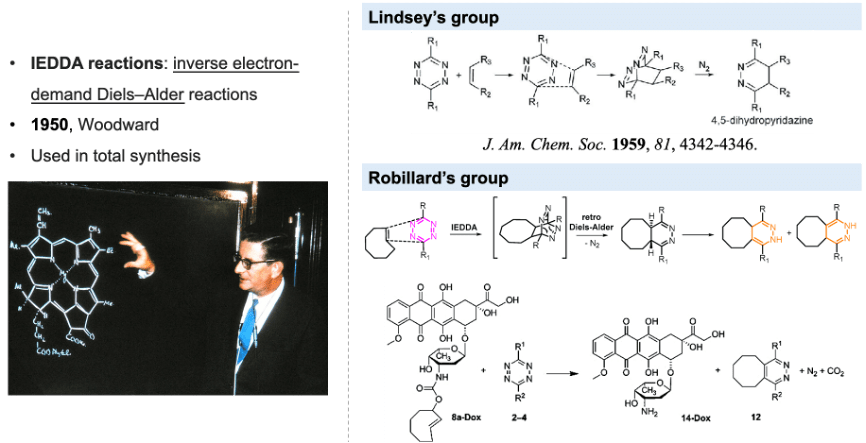
Figure 2. IEDDA reaction for drug release.
The third method is desilylation-mediated drug release. One example is the BAA-mediated deprotection of the TBS group.[6] BAA is a safe fluoride source because it has been approved by the FDA. This bioorthogonal cleavage reaction can be used in the design of antibody-drug conjugates (ADC) molecules. They conjugate BAA (Phe-BF3-) responsive linker with trastuzumab and MMAE. Then, they labeled trastuzumab with 89Zr and BAA with 18F. Mice were injected with HER2-positive BGC823 cells, and PET images show that BAA and the ADC molecule were both distributed in the tumor site. They compared the MMAE concentration in the tumor site and found that the addition of BAA can improve MMAE release, showing a significantly higher MMAE concentration compared with the group without BAA.
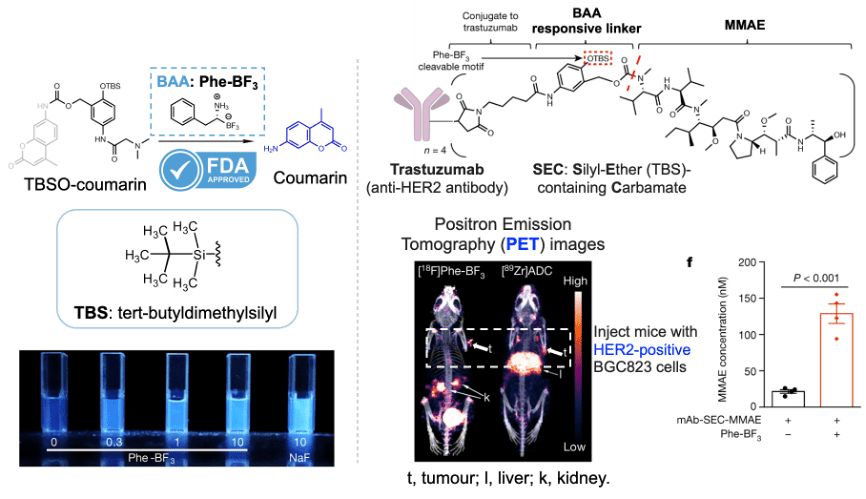
Figure 3. Desilylation for drug release.
The last method is metal-catalyzed drug activation. This type of reaction can also be used in cleavage reactions to remove the protected group and release the drug. These reactions are mainly based on the Tsuji-Trost reaction mechanism, with palladium being used as an example. First, the palladium coordinates with the alkene. The next step is oxidative addition, causing the drug to be released as a leaving group. Then, a nucleophile attacks the complex. Finally, the Pd leaves and takes part in a new catalytic cycle.
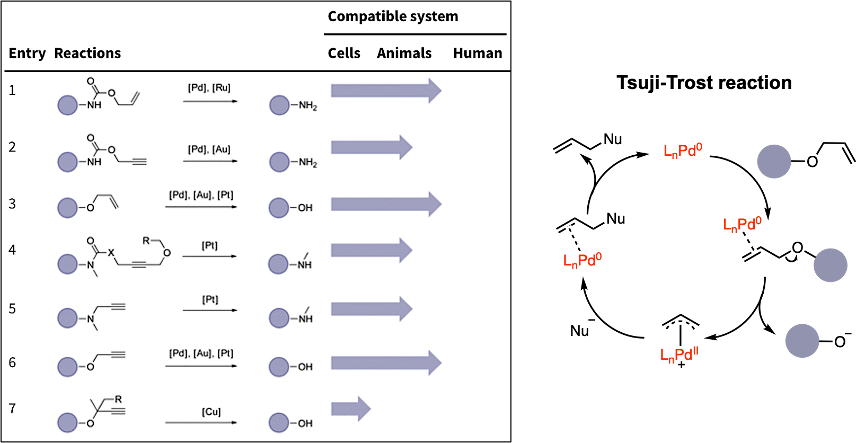
Figure 4. Metal-catalyzed cleavage reaction and mechanism of the Tsuji-Trost reaction.
In addition, metal catalysts can activate the drug through a ligation reaction. One example is the work reported by Tanaka’s Group.[7] They utilized the ring-closing metathesis (RCM) reaction to build the aromatic system. They chose the anti-tumor molecule combretastatin-A4 (CA-4) derivatives and conjugated sialic acid terminated glycan to albumin by Mannich reactions. Then, they mixed the glycan complex with ruthenium, where ruthenium binds to albumin within the hydrophobic binding pocket. Then, they injected HeLa cells into mice and, after 5 days, administered the Ru complex and compound 17 every two days for a total of 8 days. After 20 days, they measured the tumor size, and the results showed that injecting with both the Ru complex and compound 17 significantly inhibited tumor growth.
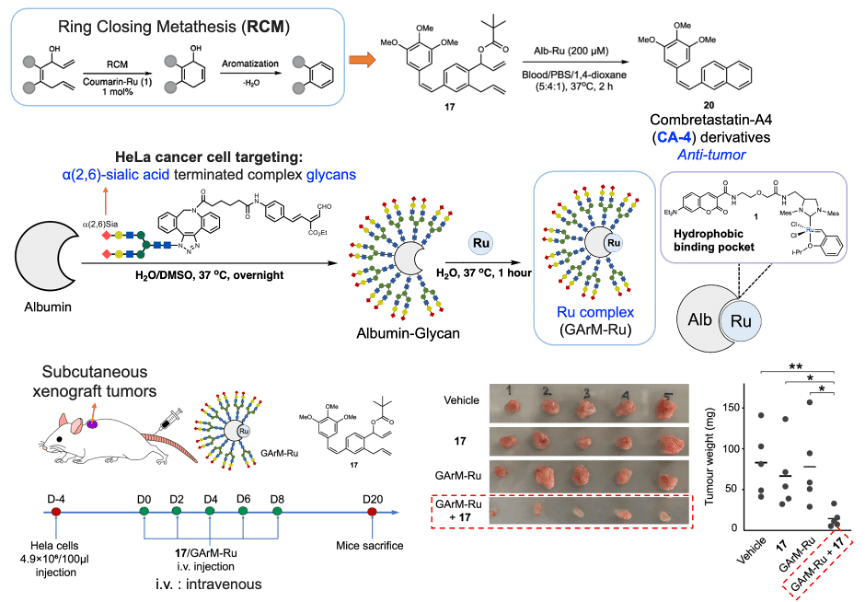
Figure 5. Synthetic prodrug design enables biocatalytic activation in mice to elicit tumor growth suppression.
In summary, numerous biorthogonal reactions have been developed for treating diseases. However, there are huge gaps between the reactions and their application due to low targeting, efficiency, and safety. More effort is needed to translate biorthogonal reactions into clinical applications.
References
- Fu, Q., Shen, S., Sun, P., Gu, Z., Bai, Y., Wang, X., Liu, Z. Chem. Soc. Rev. 2023, 52, 7737-7772.
- Azoulay, M., Tuffin, G., Sallem, W., Florent, J. C. Bioorg. Med. Chem. Lett. 2006, 16, 3147-3149.
- Matikonda, S. S., Orsi, D. L., Staudacher, V., Jenkins, I. A., Fiedler, F., Chen, J., Gamble, A. B. Chem. Sci. 2015, 6, 1212-1218.
- Versteegen, R. M., Rossin, R., ten Hoeve, W., Janssen, H. M., Robillard, M. S. Angew. Chem. Int. Ed. 2013, 52, 14112-14116.
- Liu, L., Zhang, D., Johnson, M., Devaraj, N. K. Nat. Chem. 2022, 14, 1078-1085.
- Wang, Q., Wang, Y., Ding, J., Wang, C., Zhou, X., Gao, W., Huang, H., Shao, F., Liu, Z. Nature 2020, 579, 421-426.
- Nasibullin, I., Smirnov, I., Ahmadi, P., Vong, K., Kurbangalieva, A., Tanaka, K. Nat. Commun. 2022, 13, 39.
Semi-Artificial Photosynthetic Biohybrids for Solar-Driven Green Bio-manufacturing: Opportunities and Challenges
XIAO Kemeng, 20 May 2023
Solar energy is a widely available “carbon-neutral” clean energy source in nature. Developing and popularizing bio-manufacturing technologies driven by solar energy is of great significance for addressing energy and environmental challenges of fossil fuel combustion and finally achieving the dual carbon goals. Photosynthetic organisms naturally evolve the ability to capture and covert solar energy to biomass drive material synthesis, which is the basis of life on Earth and the source of most renewable energy. However, photosynthetic organisms prioritize the optimization of survival strategies rather than solar-to-biomass conversion efficiencies, suffering limitations of low efficiency (< 6% maximum, 1-2% typical for crops and 0.1% for most other plants), photodamage, photorespiration, photoinhibition, etc.[1] Therefore, it is still challenging to achieve efficient solar-driven green bio-manufacturing.
Driven by the need to overcome the intrinsic limitations of natural and artificial photosynthetic systems, the semi-artificial photosynthetic system (SAPS) has emerged as a new type of photosynthetic system and has been evolving rapidly in recent years. This system excels in many aspects by integrating advantages from existing natural and artificial photosynthetic systems, such as achieving much better solar-utilization capability of natural systems by combining with semiconducting materials, breaking down the limitation of artificial systems to achieve the diverse range and high value of products enabled by natural biocatalysts; and capable of utilizing a broad spectrum of biological systems including the native photosynthetic species, non-photosynthetic organisms and in vitro enzymes, thus creating ample opportunities to construct novel pathways for solar-to-chemical conversion.[2] The characteristic features of natural photosynthesis, artificial photosynthesis, and semi-artificial photosynthesis are illustrated in Figure 1.[3]
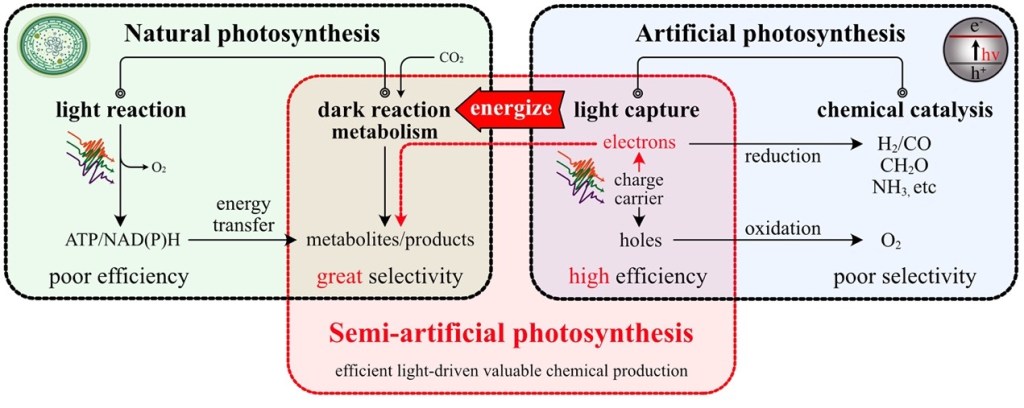
Figure 1. Schematic illustration of the basic characteristics of natural photosynthesis, artificial photosynthesis, and semi-artificial photosynthesis.[3]
The photosynthetic nano-cell hybrids have been demonstrated with great potential for efficient solar-to-chemical conversion. However, most of the current examples are essentially based on the proof-of-concept demonstrations with a limited number of chemical products under laboratory conditions. Practical application of the current hybrid systems is largely hampered due to relatively low solar-to-chemical efficiency (Figure 2). The fundamental issue for the low conversion efficiency lies in insufficient understanding of the mechanisms of electrons transfer between the biotic-abiotic interface, which is often determined by the specific localization of nanomaterials relative to the cells. Nanomaterials can be located at either extracellular space (segregated from or adhered to cell membranes) or intracellular space (located in periplasm or cytoplasm) of the target cells. Therefore, depending on specific localization of nanomaterials relative to the target cells, corresponding energy delivery mechanisms for different types of nano-cell hybrids are discussed.
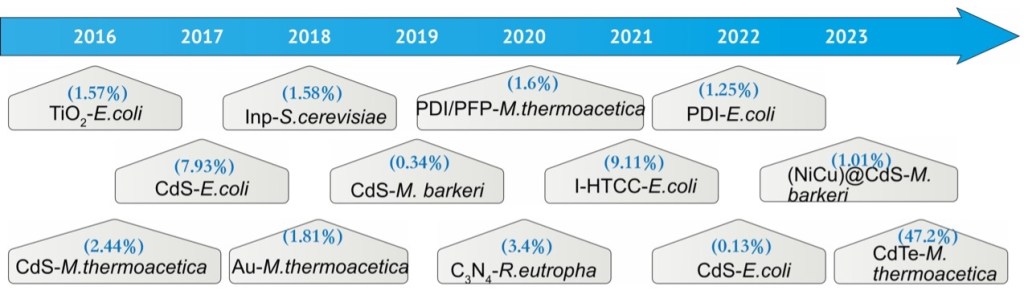
Figure 2. Solar-to-chemical conversion efficiency of typical SAPS.
Nanomaterials localized at the extracellular space of cells
When nanomaterials and microbial cells are randomly suspended in aqueous media under constant shaking (referred to as the segregated mode), the segregated nanomaterials can interact with microbial cells via random collision, during which the photoelectrons may transfer across membrane via either electron conduits or certain mediators. Redox protein complexes, often located on the plasma membrane of certain electroactive bacteria (e. g, OmcA, MtrA/B/C, and CymA, of S. oneidensis; OmcB of G. sulfurreducens), can function as electron conduits. Alternatively, the electrons generated from the nanomaterials can also transfer across cell membrane to reach the intracellular metabolic networks via the cross-membrane diffusion of certain mediators. In this case, the mediators such as the methyl viologen (MV), flavins and neutral red (NR), act as redox shuttles, which can reversibly load and unload electrons via specific redox reactions. Typical examples under the segregated mode include TiO2-C. butyricum and TiO2-E. coli (engineered to express recombinant [FeFe]-hydrogenase and maturases), both of which utilized MV as electron mediator (Figure 3).[4]
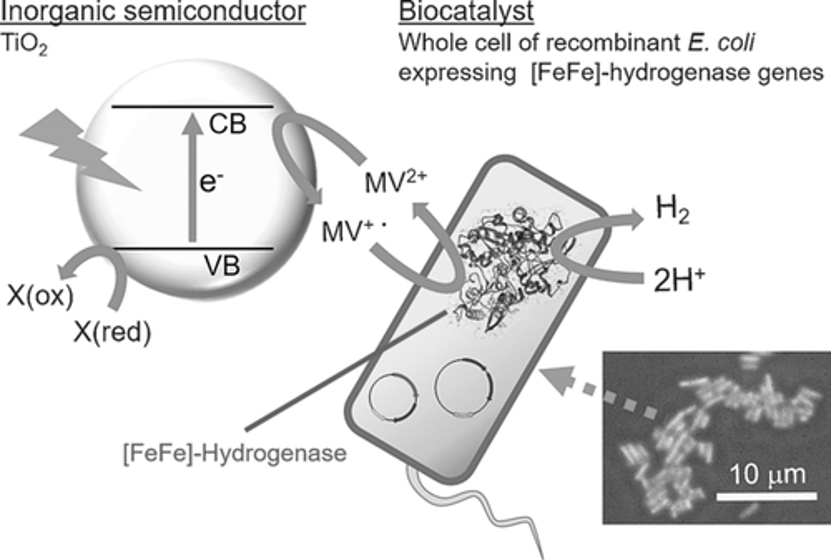
Figure 3. A typical example of segregated mode, in which TiO2 was suspended in the medium and random contacted with bacterial cells. Photocatalytic H2 production by the combination of TiO2, MV, and the recombinant E. coli.[4b]
Through physiochemical or biological approaches, nanomaterials could also be directly adhered to the surfaces of bacterial cells for the construction of nano-cell hybrids. In this mode, the persistent contact between nanomaterial and cell surface, or membrane-associated matters (e.g., surface-adhered molecules, biofilm, or metal-binding proteins), would substantially improve the opportunity of transferring generated photoelectrons across membrane via electron conduits or the diffusion of mediators. Representative examples utilizing mediators as redox shuttles under this mode include a number of CdS-E. coli systems (e.g., MV as electron shuttle and engineered E. coli with surface-displayed Cd-binding protein, and E. coli with nano-CdS deposited in biofilm as living bio-abiotic interfaces), and the CdS-C. autoethanogenum system (Fe/FMN as potential electron shuttles).[5] Except for those two-way mediators, certain one-way mediator (e.g., H2), which was photocatalytically generated by nanomaterials and then successfully delivered to intracellular enzymes, had also been reported to perform similar functions. Typical examples include the CdS-M. thermoacetica system, in which H2 was confirmed to serve as an electron mediator (Figure 4).[6]
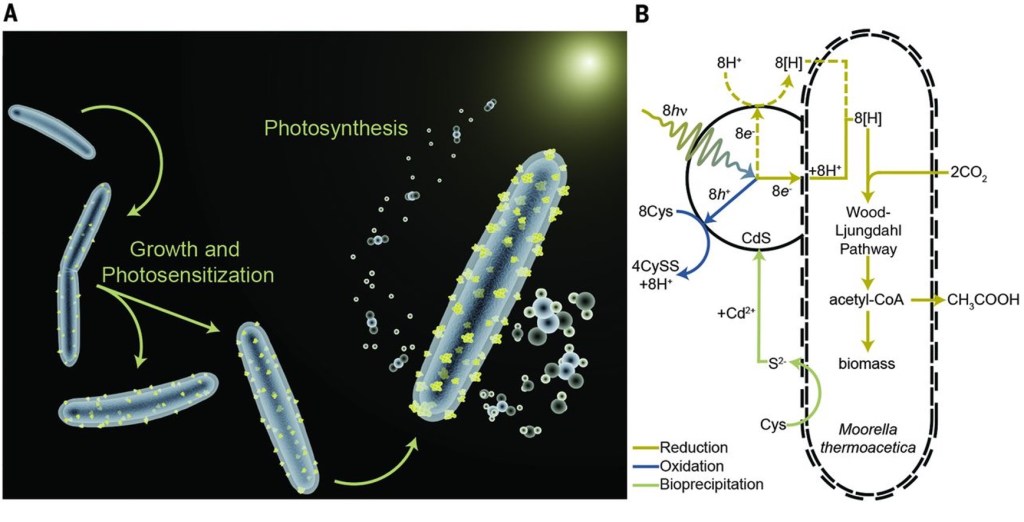
Figure 4. A typical example of surface-adhered mode, in which CdS was adhered on the surface membrane of bacterial cells. Photocatalytic acetate production by the M. thermoacetica-CdS hybrid with the H2 as mediator.[6]
For nano-cell hybrids based on organic semiconductors and microalgae, e.g., PFP-Synechococcus sp. PCC7942, photoelectrons are believed to delivered to the microbial cells via photosynthetic electron transduction chain.[7] As the corresponding redox proteins are located on thylakoid membrane rather than the cell membrane of microalgae, the actual underlying mechanisms were not completely understood. Alternative mechanisms are proposed that the organic semiconductor nanosheets may be capable of penetrating cell membrane and correspondingly deliver the photoelectrons into cytoplasm.
Nanomaterials localized inside the cells
Nanomaterials of smaller size (i.e., quantum dots, QDs) could directly penetrate into the periplasm or cytoplasm of microbial cells via endocytosis. In this scenario, the electron flow from the solar-capture nanomaterials to the intracellular metabolic networks is straightforward and is expected to feature less energy loss during transport process than the case of the nanomaterials localized outside the cells relying on transmembrane energy transport. The representative nano-cell hybrid systems containing internalized nanoparticles include Au-M. thermoacetica, CuInS2@ZnS-S. oneidensis, g-C3N4@E. coli, CdS/CdSe/InP/Cu2ZnSnS4@ZnS -A. vinelandii, and CdTe-X. autotrophicus.[8] In the Au-M. thermoacetica hybrid system, the energy delivery is believed to rely on the interaction between cytoplasm-located Au QDs and hydrogenases of Wood-Ljungdahl pathway, either via direct electron transduction or unidentified intracellular mediators (Figure 5). In the CuInS2@ZnS-S. oneidensis hybrid system, CuInS2@ZnS QDs were first taken up into the periplasm of microbial cells and the generated photoelectrons were directly delivered to periplasmic hydrogenases. Likewise, in the g-C3N4@E. coli system, the g-C3N4 QDs were first internalized by E. coli and corresponding energy were eventually transported to hydrogenases. Notably, in the CdS/CdSe/InP/Cu2ZnSnS4@ZnS-A.vinelandii system, photoelectrons generated from the cytoplasm-located CdS/CdSe/InP/Cu2ZnSnS4@ZnS QDs could be delivered to nitrogenase with high specificity due to the Zn-affinity His-tag motif that have been engineered on enzymes.
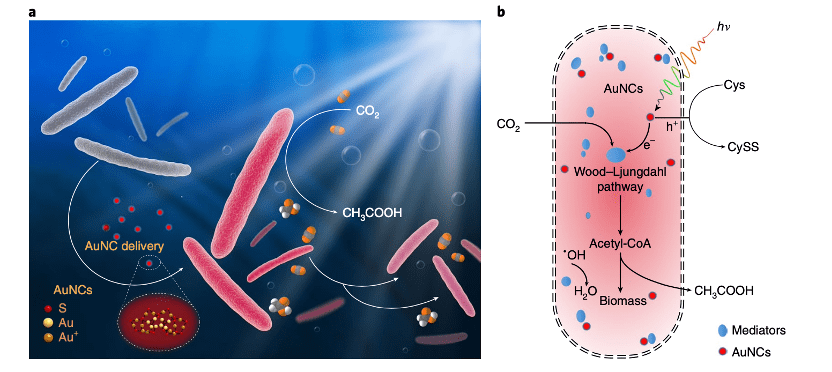
Figure 5. A typical example of uptake of QDs by bacterial cells. The Au22(SG)18 nanoclusters were delivered into bare M. thermoacetica (grey) during the culture process, forming M. thermoacetica/AuNCs with red emission. The electrons generated from intracellular AuNCs under illumination are used by enzymatic mediators inside the cytoplasm and are finally passed on to the Wood-Ljungdahl pathway.[8a]
However, most of the mechanisms of energy transport discussed above are still hypotheses that require advanced analytical techniques to reveal the underlying mechanisms. Nonetheless, the solar energy captured by extracellular localized nanomaterial undergoes multistep transformation during the cross-membrane transfer process before entering the cell, and collateral energy loss will occur at each step of transformation, which is a defect of the extracellular localized nanomaterial. A deep molecular understanding of the interactions involving the semiconductor, the bacterium, and light will further enable a guided search through the vast microbial and enzymatic parameter space to increase the efficiency and performance of such hybrid solar-to- chemical platforms, therefore promoting the green-biomanufacturing of value-added chemicals, fertilizers, and fuels from photoactivation of simple building blocks like CO2, N2 and H2O.
References
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang, E. Reisner, Nature nanotechnology 2018, 13, 890.
- S. Cestellos-Blanco, H. Zhang, J. M. Kim, Y.-x. Shen, P. Yang, Nature Catalysis 2020, 3, 245.
- K. Xiao, J. Liang, X. Wang, T. Hou, X. Ren, P. Yin, Z. Ma, C. Zeng, X. Gao, T. Yu, Energy & Environmental Science 2022, 15, 529.
- a) A. Krasnovsky, V. Nikandrov, FEBS letters 1987, 219, 93; b) Y. Honda, H. Hagiwara, S. Ida, T. Ishihara, Angewandte Chemie 2016, 128, 8177.
- a) W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu, J. Zhao, Science advances 2018, 4, eaap9253; b) X. Wang, J. Zhang, K. Li, B. An, Y. Wang, C. Zhong, Science Advances 2022, 8, eabm7665; c) S. Jin, Y. Jeon, M. S. Jeon, J. Shin, Y. Song, S. Kang, J. Bae, S. Cho, J.-K. Lee, D. R. Kim, Proceedings of the National Academy of Sciences 2021, 118, e2020552118.
- K. K. Sakimoto, A. B. Wong, P. Yang, Science 2016, 351, 74.
- Y. Zeng, X. Zhou, R. Qi, N. Dai, X. Fu, H. Zhao, K. Peng, H. Yuan, Y. Huang, F. Lv, Advanced Functional Materials 2021, 31, 2007814.
- a) H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto, P. Yang, Nature nanotechnology 2018, 13, 900; b) Y. Ding, J. R. Bertram, C. Eckert, R. R. Bommareddy, R. Patel, A. Conradie, S. Bryan, P. Nagpal, Journal of the American Chemical Society 2019, 141, 10272; c) S. Koh, Y. Choi, I. Lee, G.-M. Kim, J. Kim, Y.-S. Park, S. Y. Lee, D. C. Lee, Journal of the American Chemical Society 2022, 144, 10798; d) D. Wu, W. Zhang, B. Fu, Z. Zhang, Joule 2022, 6, 2293; e) X. Guan, S. Erşan, X. Hu, T. L. Atallah, Y. Xie, S. Lu, B. Cao, J. Sun, K. Wu, Y. Huang, Nature Catalysis 2022, 5, 1019.
Narcolepsy
LIU Min, 16 March 2023
Narcolepsy is a neurological disorder that affects the brain’s ability to regulate sleep-wake cycles. Individuals with narcolepsy experience excessive daytime sleepiness and may suddenly and uncontrollably fall asleep at any time during the day, even in the middle of an activity. Additionally, they may experience sudden episodes of muscle weakness or paralysis, known as cataplexy, triggered by strong emotions. Other symptoms of narcolepsy may include vivid dreams, sleep paralysis, and hallucinations upon falling asleep or waking up. There are two main types of narcolepsy: NT1, which is typically with cataplexy, orexin levels are low. And NT2, which is without cataplexy, orexin levels are normal.
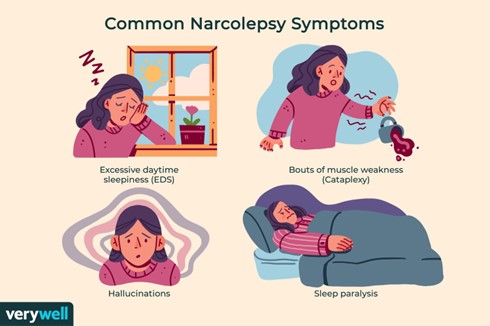
The cause of narcolepsy is a deficiency of hypocretin, a neurotransmitter produced by cells in the brain that help regulate the sleep-wake cycle. This discovery has revolutionized the treatment of narcolepsy and has also helped to reduce the stigma associated with the disorder. Orexin is a polypeptide produced by neurons in the hypothalamus. It plays a critical role in regulating sleep and wakefulness, as well as appetite, energy metabolism, and reward-seeking behavior. Orexin is released when the body is in a wakeful state, promoting wakefulness and regulating the sleep-wake cycle. It is found in a pair of hypothalamic nuclei in the brain and affects various processes, including arousal, appetite, and sleep. Deficiency of orexin has been linked to narcolepsy, a disorder characterized by excessive daytime sleepiness and sudden sleep attacks. Additionally, orexin has been implicated in addiction and mood disorders. The loss of hypocretin can lead to problems in controlling the sleep-wake cycle, resulting in excessive daytime sleepiness and sudden sleep attacks, as well as other symptoms such as cataplexy, sleep paralysis, and hallucinations.
While the exact cause of the cell loss is not fully understood, it is believed to be a combination of genetic and environmental factors. There is no known cure for narcolepsy, but it can be managed with medications and lifestyle changes. Treatment may include stimulant medications to combat daytime sleepiness, antidepressants to alleviate cataplexy and other symptoms, and lifestyle changes such as maintaining a regular sleep schedule, avoiding caffeine and alcohol, and taking short naps during the day. The main treatment of this disease contains the following parts. The first one is stimulants, such as modafinil, can block several monoaminergic transporters, the mechanism underlying the wake-enhancing function of this drug is probably mainly due to dopamine reuptake inhibition, through blockage of the dopamine transporter The second one is sodium oxybate, which activates the GABA type B receptor and possibly its own specific GHB receptor. It is an existing depression and untreated sleep disease. In addition, there are off-label to treat cataplexy, such as antidepressants, which can reduce cataplexy quickly (often within a few hours), but the abrupt withdrawal of treatment can result in a severe worsening of cataplexy.
The current challenges in restoring orexin signaling are that the natural orexin molecules are not suitable. If taken orally, orexins would be broken down quickly in the gut. If given intravenously, only 1% would actually get into the brain. Injection into the brain should work, but that has many problems. Controversial whether orexins can get into the brain from the nasal lining. And all these methods carry the risk of an autoimmune response to orexin peptides. So, the goal is to design a small molecule that can mimic the effects of orexins. The main nomenclature is Ox2R agonist, and its drug binds to and activates the Ox2 receptor. And the other Ox2R antagonist is the drug that binds to and blocks the Ox2R receptor.
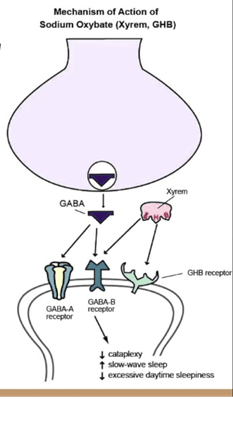
References
- Kornum, B. R., Knudsen, S., Ollila, H. M., Pizza, F., Jennum, P. J., Dauvilliers, Y., & Overeem, S. (2017). Narcolepsy. Nature reviews Disease primers, 3(1), 1-19.
- Bassetti, C. L., Adamantidis, A., Burdakov, D., Han, F., Gay, S., Kallweit, U., … & Dauvilliers, Y. (2019). Narcolepsy—clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nature Reviews Neurology, 15(9), 519-539.
- Siegel, J. M. (1999). Narcolepsy: a key role for hypocretins (orexins). Cell, 98(4), 409-412.
- Akintomide, G. S., & Rickards, H. (2011). Narcolepsy: a review. Neuropsychiatric disease and treatment, 507-518.
Understanding the Electron Transfer Pathway of Cytochrome P450s: A Structural Point of View
WANG Yue, 2 March 2023
Cytochrome P450s are a large and ancient enzyme superfamily found in all kingdoms of life. They have a unique ability to detoxify chemicals in humans, making them an important target in medicinal and pharmaceutical drug discovery. The term “P450” is derived from the spectrophotometric peak at the wavelength of the absorption maximum of the enzyme (450 nm) when it is in the reduced state and complexed with carbon monoxide. The official nomenclature of P450 is based on gene sequence similarity, which assigns a family number (e.g., CYP1, CYP2), followed by a subfamily letter (e.g., CYP1A, CYP2B) and then several individual proteins (e.g., CYP1A1, CYP2B6).
P450 systems mainly consist of two functional parts: the heme-containing P450 domain and redox partners (RPs). The P450 domain contributes to substrate binding and transformation, while the RPs contain redox centers that relay electron equivalents from electron donors (usually NADPH) to activate the dioxygen bound to the P450 domain. Together, these events form an electron transfer (ET) pathway of NADPH-RPs-heme-O2.
Coacervates are condensed liquid-like droplets formed by liquid–liquid phase separation of molecules through multiple weak associative interactions. In recent years it has emerged that not only long polymers, but also short peptides can form simple and complex coacervates. This emerging peptide-based coacervate holds promise as efficacious delivery vehicles for multi-purpose biomedical applications.
The core region of the heme domain of all known P450s folds into a triangular globular domain (Figure 1A). Membrane-bound P450s have an additional N-terminal transmembrane domain and a flexible linker that may influence the localization and orientation in the membrane. The catalytic center of P450 is a Fe ion that is ligated to a protoporphyrin XI macrocycle and two additional axial ligands[1]. The proximal one is a thiolate of the strictly conserved Cys from the enzyme, and the distal one is a ligand that varies during the catalytic cycle. The elongated helix I running through the distal side of the heme contains residues that interact with substrate and dioxygen (Figure 1A). Helix F and G, which lie above helix I, are highly flexible and control the opening of the substrate entry tunnel.[2] The space of the closed-form pocket varies substantially in different P450 domains, determining the enzyme-substrate specificity. The heme-coordinating Cys residue located on the C-terminus of the helix L is situated on the proximal side of the heme group (Figure 1B). The proximal side is considered a portal to accept electrons delivered from RPs, and thus there are fewer structural elements on this side. The Cys sulfur is hydrogen-bonded to the nearby peptide NH groups to render the heme iron a sufficient redox potential.[3-4]
Because P450 domains share a similar fold, using the diverse RP systems used by various P450s is also a common approach to categorize these enzymes. Class II P450s are the most common eukaryotic P450s and are generally found in the endoplasmic reticulum (ER). These P450s employ a two-component system that comprises a membranous P450 domain and NADPH-cytochrome P450 reductase (CPR; Figure 2, class II). In the human liver, class II CYPs and CPRs are anchored on ER membrane by a single TM domain with their catalytic domain facing the cytosol. CPR contains FAD-containing reductase and FMN-binding domains that are linked via a “hinge” region, which is presumed to confer conformational plasticity of two flavin-binding domains.
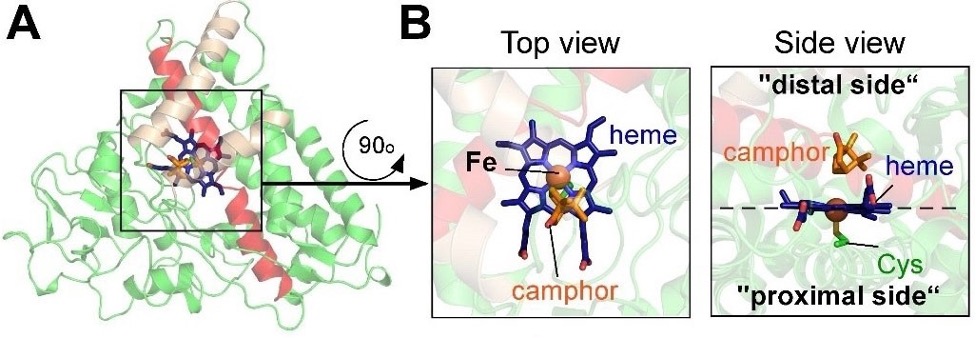
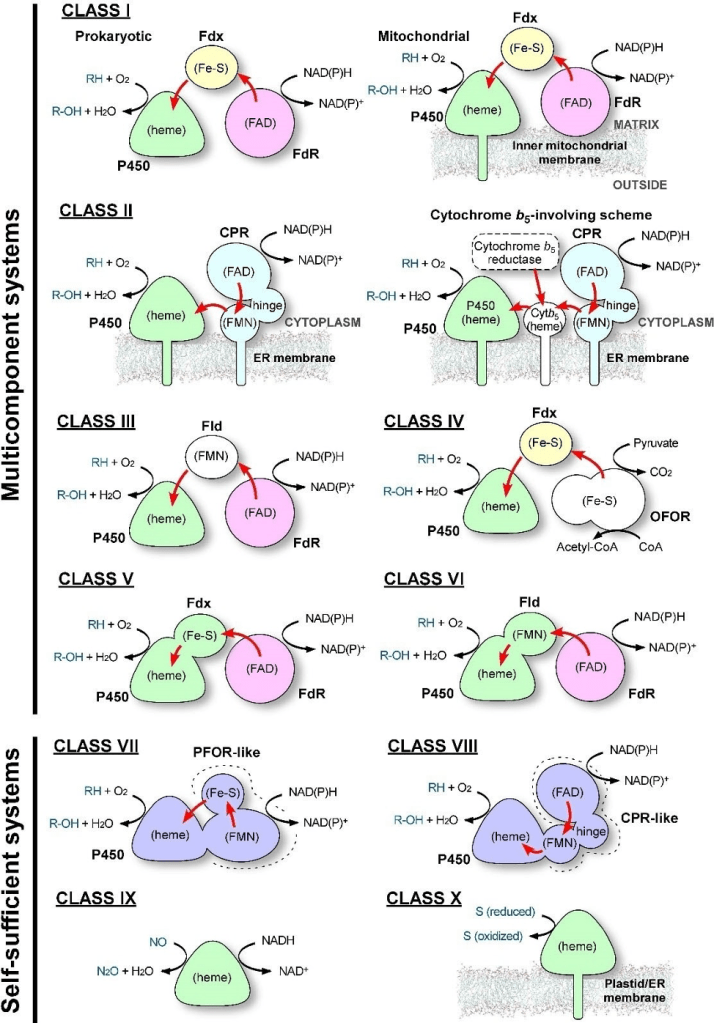
References
- Chen, C. C., Min, J., Zhang, L., Yang, Y., Yu, X., & Guo, R. T. ChemBioChem 2021, 22, 8, 1317-1328.
- Y.-T. Lee, R. F. Wilson, I. Rupniewski, D. B. Goodin, Biochemistry 2010, 49, 3412–3419.
- a) R. Langen, G. M. Jensen, U. Jacob, P. J. Stephens, A. Warshel, J. Biol. Chem. 1992, 267, 25625–25627.
- P. J. Stephens, D. R. Jollie, A. Warshel, Chem. Rev. 1996, 96, 2491–2514.
- R. Bernhardt, V. B. Urlacher, Appl. Microbiol. Biotechnol. 2014, 98, 61856203.
Peptide-based liquid droplets as emerging delivery vehicles
CHEN Hongfei, 2 Feb 2023
Coacervates are condensed liquid-like droplets formed by liquid–liquid phase separation of molecules through multiple weak associative interactions. In recent years it has emerged that not only long polymers, but also short peptides can form simple and complex coacervates. This emerging peptide-based coacervate holds promise as efficacious delivery vehicles for multi-purpose biomedical applications.
1. Peptides as building blocks for liquid microdroplets
There are mainly four kinds of peptide-based coacervate: simple coacervate formed by peptides and peptide derivatives, polymers formed by repeated peptide motifs, complex of oppositely charged peptides and polyelectrolytes, and polymer–peptide hybrids (Figure 1). [1-4] Simple coacervation of peptides is formed by interactions of a single peptide or peptide derivative in aqueous buffer which often triggered by a change in temperature, pH or salinity. The microdroplets contain most of the peptides originally in solution and a significant amount of hydration water. [1] The engineering peptide polymers containing tandem repeats of sequences can form coacervate more easily. Compared with short peptides, peptide polymers are more prone to phase separation because their mixing entropy per amino acid is smaller. [2] By combining two oppositely charged peptides complex coacervates can be formed. Oligopeptides as short as ten amino acids can form stable salt-responsive complex coacervate microdroplets whose critical salt concentrations can be tuned over two orders of magnitude by changing peptide length and composition. [3] Besides of this, short peptides can be grafted onto a hydrophilic polymer backbone to enhance their hydration and create coacervates that more closely mimic the physicochemical properties of membraneless organelles. The hybrids phase separates into liquid microdroplets resembling membraneless organelles in vitro and in living cells. [4]
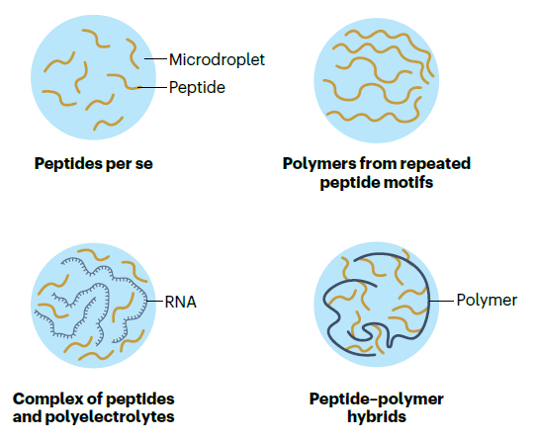
2. Peptide-based coacervates as stimuli-responsive delivery vehicles
Peptide-based coacervate droplets, which range from nanometers to micrometers, are promising delivery vehicles. Within the microdroplets, molecules can be hold by weak non-covalent interactions, allowing the packaging of different types of therapeutics. Compared with solid particles that are more commonly used for delivery, such as polymer, inorganic or lipid nanoparticles, coacervates are viscoelastic objects that can quickly recruit cargoes under aqueous conditions, which is especially beneficial for preserving the bioactivity of biomacromolecular therapeutics (Figure 2).
Coacervate, developed from thermo-responsive ELPs fused to therapeutics, can greatly prolong retention times of the therapeutics when injected subcutaneously or directly at the tumor sites. Rational molecular design can tune the coacervation temperature which enables the peptides to be injected as a solution at room temperature and then form viscous depots at body temperature. Using this concept, an ELP fused to glucagonlike peptide 1 (a promising drug for treatment of type 2 diabetes) was designed so that it was soluble at ambient temperature for ease of injection, but quickly transitioned at body temperature into a droplet with slow-releasing characteristics. With this strategy, the blood sugar level could be controlled for up to 17 days in a diabetic primate animal model. [5] Peptide microdroplets can also be used to deliver therapeutics inside the cell.
A cationic peptide was designed to form complex coacervates with the antibody immunoglobulin G. This kind of coacervate was able to cross the cell membrane and deliver immunoglobulin G inside the cell, opening interesting possibilities for antibody therapies. [6] These examples establish the molecular versatility afforded by peptides to form therapeutic containing microdroplets.
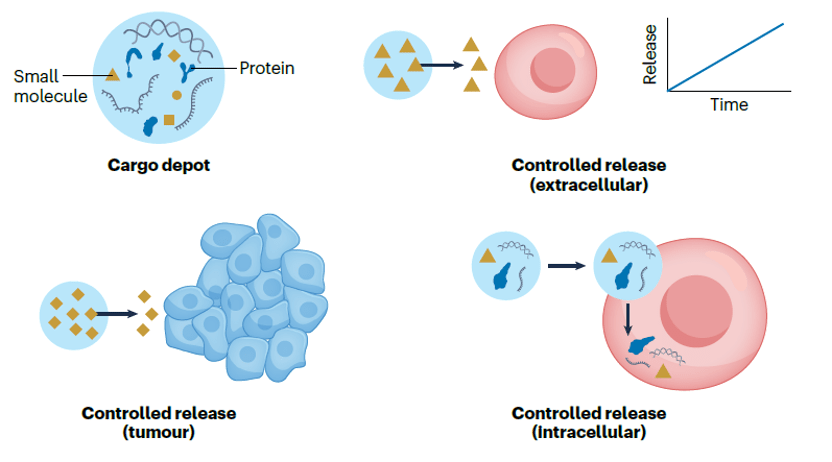
3. Outlook
Peptide-based microdroplets create new opportunities for delivery compared with other types of delivery vehicles. The formation of these microdroplets is nearly instantaneous and does not require organic solvents that can reduce the bioactivity of entrapped molecules. A key advantage over other delivery vehicles is their relative simplicity of formulation and their safety in use.
Nonetheless, there are challenges. Liquid droplets may be less stable than solid particles, especially against dilution, and may disassemble prior to reaching their target. They are also prone to fusing over time in storage. Although it is known that coacervates can cross the cell membrane, it is not yet clear how they do so: it is necessary and challenging to understand the cellular uptake pathways.
References
- Abbas, M., Lipiński, W. P., Nakashima, K. K., Huck, W. T. S. & Spruijt, E. A short peptide synthon for liquid–liquid phase separation. Nat. Chem. 13, 1046–1054 (2021).
- Simon, J. R., Carroll, N. J., Rubinstein, M., Chilkoti, A. & López, G. P. Programming
molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat. Chem. 9, 509–515 (2017). - Cakmak, F. P., Choi, S., Meyer, M. O., Bevilacqua, P. C. & Keating, C. D. Prebiotically-relevant low polyion multivalency can improve functionality of membraneless compartments. Nat. Commun. 11, 5949 (2020).
- Liu, J., Zhorabek, F., Dai, X., Huang, J. & Chau, Y. Minimalist design of an intrinsically disordered protein-mimicking scaffold for an artificial membraneless organelle. ACS Cent. Sci. 8, 493–500 (2022).
- Luginbuhl, K. M. et al. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 1, 0078 (2017).
- Iwata, T. et al. Liquid droplet formation and facile cytosolic translocation of IgG in the presence of attenuated cationic amphiphilic lytic peptides. Angew. Chem. Int. Ed. 60, 19804–19812 (2021).
Introduction to Antibody-Drug Conjugates and Recent Advances in Linker
LI Biquan 22 Dec 2022
Antibody-drug conjugates or ADCs are a class of biopharmaceutical drugs designed as a targeted therapy for treating cancer. ADCs combine the targeting properties of monoclonal antibodies with the cancer-killing capabilities of cytotoxic drugs, designed to discriminate between healthy and diseased tissue[1]. Totally, ADCs include three parts: antibody, linker and drug, as shown in Figure 1. Firstly, we could divide the conjugations between antibody and linker to site-specific conjugations and non-specific conjugations. On the other hand, linkers comprise two key parts: the antibody- and the payload-attachment (Figure 1). The bonding between the linker and the mAb is critical since it defines the drug-to-antibody- ratio (DAR, in generally 3 to 4 and maximum would not exceed 8 to 9) and therefore the homogeneity and stability of the ADC. The bonding between the linker and the payload is equally important and determine the releasing mechanisms of the drugs[2-4].
Additionally, we have to make a clear mind at the mechanisms that how ADCs enter the target cells. As shown in Figure 2 (A), endocytosis leads ADCs to the lysosome, a unique compartment with a low pH and high concentration of hydrolytic enzymes. Alternatively, ADC linkers can also be labile to the extracellular tumor microenvironment, according to the non-internalizing mechanism of action previously discussed (Figure 2 (B))[2-3].
For ADCs to be selective and potent, the linker technology employed should strive towards three key properties: (1) High stability in circulation. (2) High water solubility to aid bioconjugation and avoid formation of inactive ADC aggregates. (3) Allow efficient release of a highly cytotoxic payload-linker metabolite[3]. ADC linkers can be classified as ‘cleavable’ or ‘non-cleavable’. For non-cleavable linkers, there is no inbuilt chemical trigger to cleave the linker. Conversely, cleavable linkers exploit specific conditions to release the drug at the target cell. More than 80% of the clinically approved ADCs employ cleavable linkers. In addition, cleavable linkers can be further subcategorized as chemically- cleavable or enzyme-cleavable[3].
Chemically cleavable linkers could be subcategorized as acid cleavable, reducible disulfides and those cleavable by exogenous stimuli, such as extra adding chemicals and photo triggers. On the other hand, enzyme cleavable linkers so far include cathepsins cleavable, glycosidase cleavable sulfatase cleavable and phosphatase cleavable linkers[3-4].
1 Chemically Cleavable Linkers
Acid cleavable linkers aim to exploit the acidity of the endosomes (pH 5.5-6.2) and lysosomes (pH 4.5-5.0), whilst maintaining stability in circulation at pH 7.4. This strategy yielded the earliest clinical success in the field with Pfizer’s gemtuzumab ozogamicin (Mylotarg) ADC and was later employed in Besponsa[5-6] (Shown in Figure 3). In 2019, the group[7] developed a novel silyl ether-based acid-cleavable ADC carrying highly cytotoxic monomethyl auristatin E (Shown in Figure 3). Glutathione (GSH)-cleavable (reducible disulfides) linkers rely on the higher level of glutathione in the cytoplasm (1-10 mmol/L) compared to the blood plasma (5 mmol/L)[8]. Disulfides are the most prominent class of chemically cleavable motifs found in ADC linkers and, alongside hydrazones, they have found clinical success in Pfizer’s Mylotarg and Besponsa. Disulfides are stable at physiologic pH but are susceptible to nucleophilic attack from thiols (Figure 4) [3-4]. The examples are shown in Figure 4. Whilst the cleavage of ADC linkers with endogenous triggers is the simplest method for drug release, external small molecule triggers of extracellular drug release may offer the following advantages: (1) avoidance of any disparity in linker cleavage rates caused by variable biology across patients, and (2) the ADCs can be effective when extracellular concentrations of endogenous triggers are insufficient for efficient payload release[4].
Stenton et al. have described a thioether-containing linker that, upon exposure to Pd (0), releases an amine-linked payload (Figure 5 (A))[9]. Rossin et al. have recently developed an ADC bearing an antibody fragment and a trans-cyclooctene linker that, upon inverse-electron demand Diels–Alder (IEDDA) reaction with an externally administered tetrazine, releases an amine-containing payload in vivo (Figure 5 (B))[10].
The strategy of payload release based on photo-responsive cleavable triggers has gradually emerged in recent years. Photo-responsive cleavable triggers have the following advantages: (1) controlled drug release and effective reduction in off-target toxicity and (2) determination of the cleavage mechanism and no limitation on the intracellular release. Photo- responsive cleavable triggers enjoy the following advantages, including low toxicity, a rapid response, and high sensitivity and specificity[4].
In 2015, Nani et al. [11] firstly employed a near-infrared (NIR) light photo-caging strategy for ADCs (Figure 6 (A)). More recently, our group initially reported a novel ultraviolet (UV) light-controlled ADC (Figure 6 (B)) [12]. The linkers introduced a UV light-controlled O-nitrobenzyl group as a chemical trigger.
2 Enzyme Cleavable Linkers
In the classical mechanism of action, ADCs are trafficked to the lysosome of a cell where high concentrations of unique hydrolytic enzymes reside, thereby offering an opportunity for enzyme cleavable linkers to be selectively cleaved intracellularly. For selective payload release in the extracellular tumor area, enzymes that are upregulated in tumor microenvironments must be exploited[3].
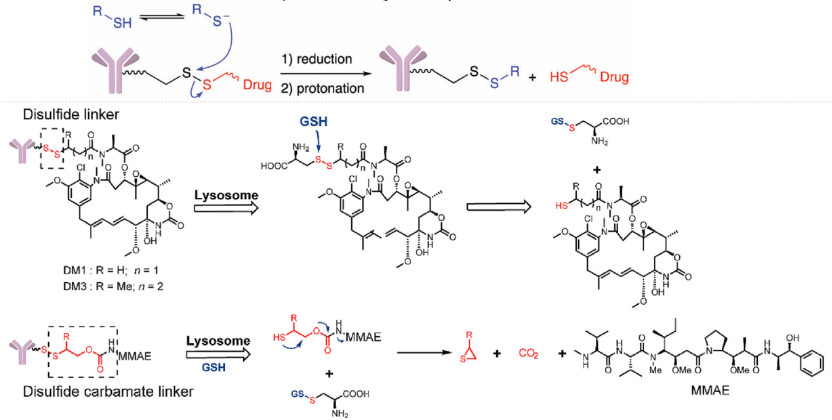
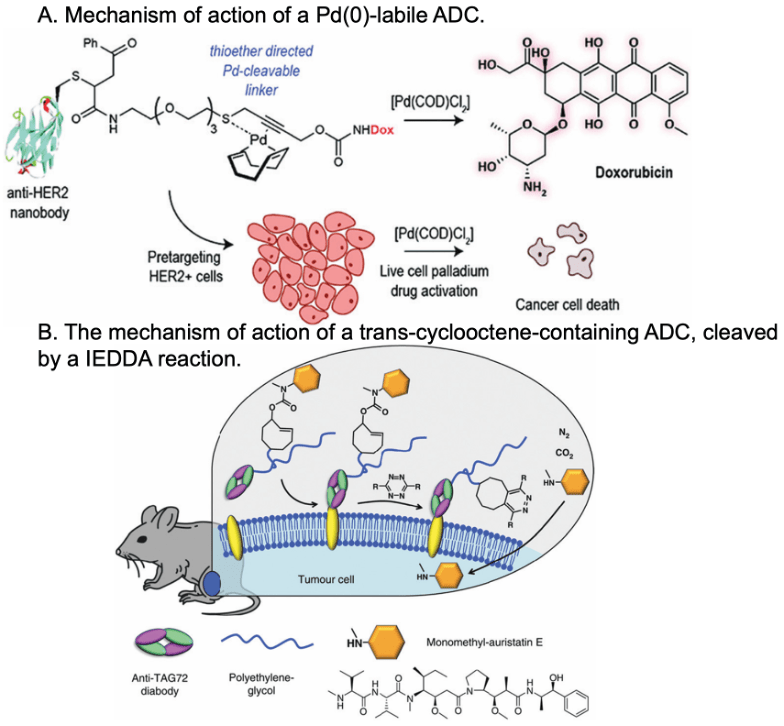
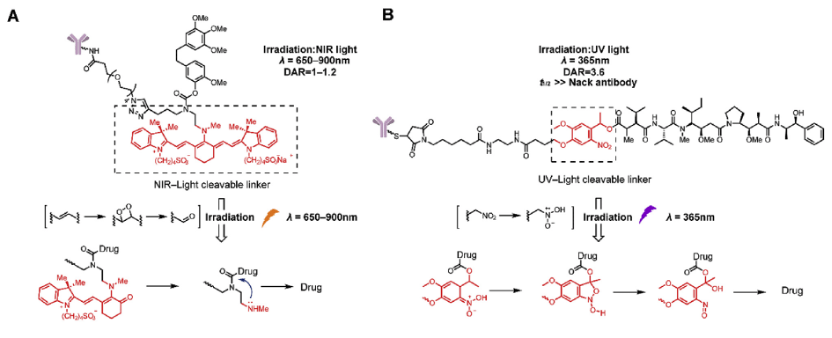
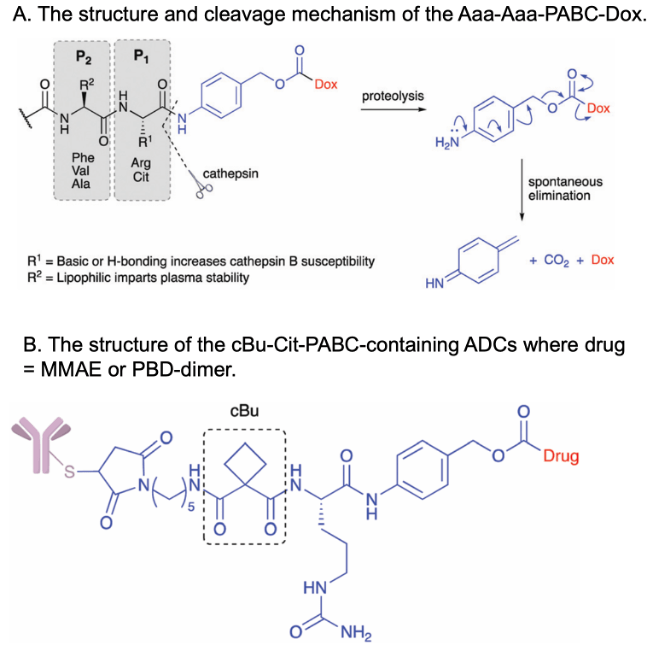
Cathepsin B is also commonly implicated in tumor progression, with overexpression and extracellular activity observed in a wide range of cancers. Cleavable dipeptides were first developed as cathepsin B-cleavable linkers for Doxorubicin (Dox)-prodrugs in 1998[3]. In 2017 Caculitan et al. [13] discovered that the valine-citrulline (Val-Cit) linker exhibited widespread sensitivity to a variety of cathepsins, including cathepsin B, cathepsin K, cathepsin L, etc. This could be detrimental, as only cathepsin B is thought to be highly expressed in cancer cells, and the widespread sensitivity to other cathepsins could induce off-target toxicity in normal cells (Figure 7 (A)). Wei et al. [14] designed a linker that used a cyclobutane-1,1- dicarboxamide (cBu) structure that was predominantly dependent on cathepsin B (Figure 7 (B)).
Similarly to cathepsins, β-glucuronidases are secreted in some tumors to necrotic areas, where they can function extracellularly. This extracellular activity led to the development of β-glucuronidase-cleavable prodrugs as early as 1988, whereby selectivity for the tumor was achieved by extracellular enzymatic cleavage in the vicinity of malignant cells (Figure 8 (A)). In addition to the classic β-glucuronidase-cleavable linkers that were developed for ADCs in 2006 [15], β-galactosidase was discovered to be overexpressed in tumor cells and possess hydrolytic activity(Figure 8 (B)). In 2017, Kolodych et al [16]. described a β-galactosidase-cleavable linker-containing ADCs (Figure 8 (C)). In 2020, a sulfatase-cleavable linker was described by Bargh et al. [17] (Figure 9). Sulfatase, which is analogous to β-galactosidase, is a hydrolytic enzyme overexpressed in tumor cells. The sulfatase-cleavable linker exhibited definite susceptibility to sulfatase enzymes (t1/2 = 24 min) in a release study.
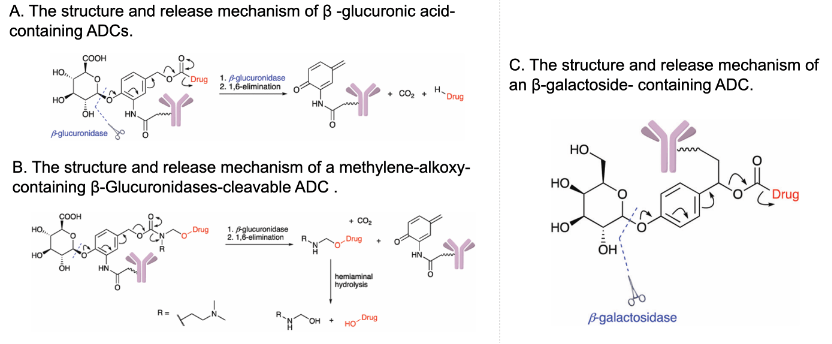
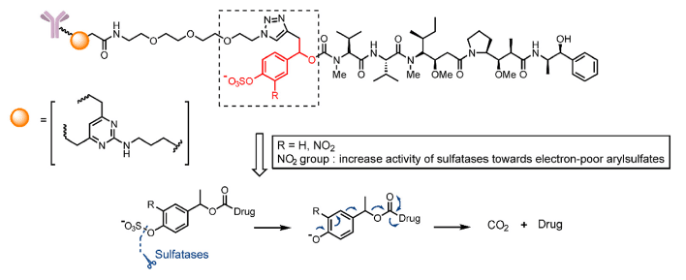

Phosphate and pyrophosphate groups can markedly improve the hydrophilicity of linkers and can be employed to load lipotropic payloads. In 2016, Kern et al. [18] employed a terminal phosphate/pyrophosphate as both a leaving group and hydrophilic group for the Val-Cit-p-aminobenzyloxycarbonyl (PAB) linker with the highly lipophilic glucocorticoid budesonide (Figure 10 (A)). It was proved that that the phosphate/pyrophosphate structure had the potential to be a novel linker. In the same year, Kern et al. [19] replaced the traditional Val-Cit-PAB linker with a phosphate diester structure and synthesized a series of linkers based on the structure of monophosphate, pyro- phosphate and triphosphate diesters (Figure 10 (B)). Compared with the previous linker, the pyrophosphate linker showed extremely high stability over 7 days in mouse and human plasma stabilization experiments.
References
[1] Biologicals, 2015, 43 (5): 318-332.
[2] Acta Pharmaceutica Sinica B, 2020, 10 (9): 1589-1600.
[3] Chem. Soc. Rev., 2019, 48, 4361-4374.
[4] Acta Pharmaceutica Sinica B, 2021, 11 (12): 3889-3907.
[5] Angew. Chem., Int. Ed., 2014, 53: 3796-3827.
[6] Bioconjugate Chem., 2015, 26: 650-659.
[7] Cancers (Basel), 2019, 11: 957.
[8] Biochem Pharmacol, 1996, 52: 401-406.
[9] Chem. Sci., 2018, 9: 4185-4189.
[10] Bioconjugate Chem., 2016, 27: 1697-1706.
[11] Angew Chem Int Ed Engl, 2015, 54: 13635-13638.[12] Bioorg Chem, 2021,111:104475.
[13] Cancer Res, 2017, 77: 7027-7037.
[14] Med Chem, 2018, 61:989-1000.
[15] Bioconjug Chem, 2006, 17: 831-840.
[16] Eur J Med Chem, 2017, 142: 376-382.
[17] Chem Sci, 2020,11: 2375-2380.
[18] Bioconjug Chem, 2016,27: 2081-2088.
[19] Am Chem Soc, 2016, 138: 1430-1445.
Peptide-based coacervates as biomimetic protocells
BAO Yishu, 24 Nov 2022
In the 1930s, Oparin first proposed that protocells could be formed by membraneless compartments, such as coacervates. Coacervate droplets are formed spontaneously by the physical phenomenon of liquid-liquid phase separation (LLPS), in which a liquid condensed phase that is typically rich in macromolecules separates from an aqueous solution. [1] This is a spontaneous process that is like the separation of oil and water, with the main difference between phase-separated coacervate systems and oil-in-water emulsions being that water is the continuous phase both inside and outside the coacervates. For this reason, coacervates have liquid properties; they can fuse, deform, and recover quickly after photobleaching. Coacervates lack a membrane, so they can easily take up and concentrate solutes from the surroundings and are prone to aggregation and precipitation (Figure 1).
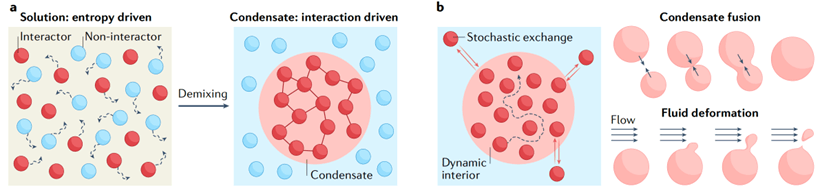
There are three types of liquid-liquid phase separation: segregative, associative, and simple phase separation (Figure 2). [2] In segregative phase separation, two soluble molecules do not mix despite a favorable mixing entropy due to repulsive interactions between them. As a result, they end up in two separate phases, each enriched in one of the solutes. A classic example of segregative phase separation is that of poly (ethylene glycol) and dextran. In associative phase separation, two soluble molecules end up in the same phase, due to attractive interactions between them. These weak interactions mainly include charge-charge interactions, dipole-dipole interactions, hydrogen bond interactions, and π-π stacking. One of the most classic examples is the phase separation of alginate (a negatively charged polysaccharide) and gelatin (a positively charged protein) in an aqueous solution. The third type is simple phase separation, whose driving forces are very similar to complex phase separation.
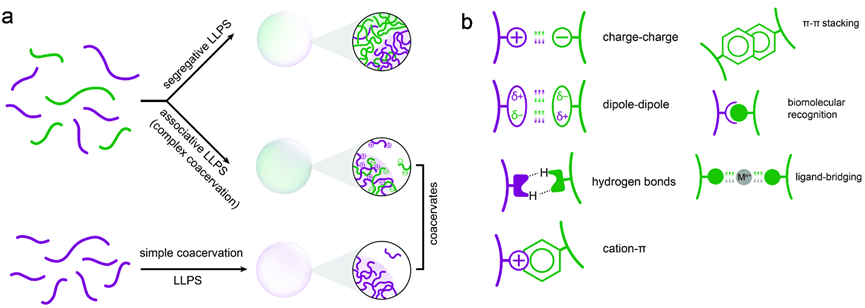
Recent studies have shown that proteins with low complexity regions (LCRs) are more prone to undergo phase separation. Therefore, one of the challenges for coacervates is to identify the simplest molecules capable of phase separating under mild conditions. Because peptides have a relatively simple structure (peptide backbone) and can introduce a variety of functional groups through side chains, they have become one of the hotspots of research. Peptide-rich coacervates are formed by associative or simple phase separation. [2]
- Complex coacervates
Complex coacervates are also divided into several types. The first type is peptide-nucleotide coacervates. In 2011, Mann and co-workers first proposed that poly-L-lysine can form coacervate microdroplets with ATP and other mononucleotides through phase separation from aqueous solutions. These droplets were stable between pH 2 and 10 and at temperatures as high as 90 oC. [3]
To improve the understanding of the interaction of peptide-nucleotide coacervates, Das and co-workers increased the complexity of the peptide sequence, and they designed Fmoc-VVVRRKK-NH2 that contains in total four cationic residues and three hydrophobic residues along with the hydrophobic Fmoc group, but this amphiphilic peptide motif cannot form coacervates with ATP at any concentration. Instead, solid nanofibers are formed due to strong charge interactions. By scrambling the amino acid positions in the peptide motif to Fmoc-KVRVRVK-NH2, the assembly was drastically altered. They found that this peptide could undergo phase separation with ATP. After the addition of ATPase, the coacervates were disassembled, indicating the reversibility of this system (Figure 3). [4]
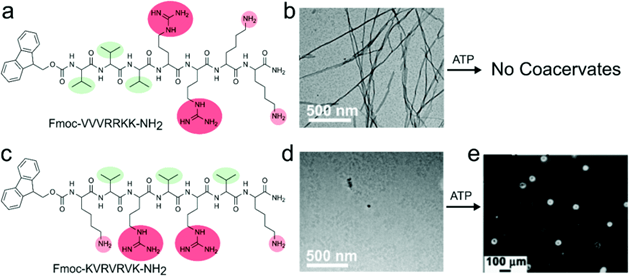
The second type is the peptide-peptide coacervates. A typical example is poly-lysine and poly-glutamate, where charge-charge interactions are thought to be the main driving force for coacervate formation. Later, Perry and co-workers found that chirality also affects the behavior of peptide-peptide phase separation. [5]
The last type is the peptide-inorganic coacervates. Li and co-workers reported a new kind of short peptide-based coacervate by condensation of cationic tripeptides with inorganic anionic polyoxometalates (POM). The main driving force behind the phase separation is thought to be the attraction between protonated amine groups and the anionic POM. [6]
2. Simple coacervates
Unlike complex coacervates, simple coacervates contain a single component that is responsible for phase separation. Underwater organisms are one of the sources of coacervate-forming proteins and peptides, such as mussel foot protein (mfp). Even though mfp-3S is a relatively short protein (45 amino acids), Wei and co-workers managed to design a Mussel foot-derived peptide, called mfp-3S-pep (25 amino acids), which expressed the ability to self-coacervate at similar conditions of pH and salt as the protein (Figure 4). [7]
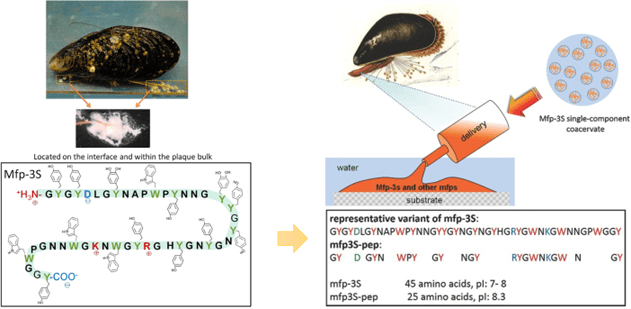
Another interesting example of a marine organism that exploits protein-based coacervates is the jumbo squid. [8] Its hard beak grows as a result of the secretion of histidine-rich proteins (histidine-rich beak proteins, HBPs) that form coacervates in seawater. In 2019, Gabryelczyk and co-workers found that GHGLY is the basic sequence motif that drives phase separation, and four repeats of this sequence is the smallest number required to trigger LLPS (Figure 5).
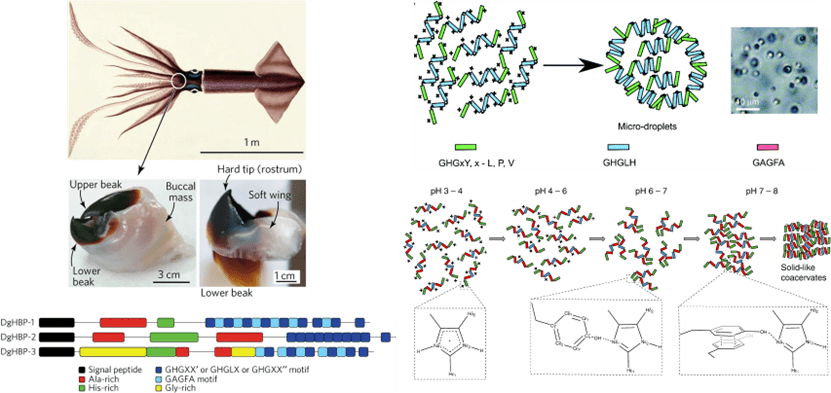
The last one is designer short peptide motifs. Evan Spruijt and co-workers designed short peptide derivatives based on the sticker and spacer model. This peptide-based coacervate can act as a microreactor in which the rates of two types of addition reactions, including aldol and hydrazone formation, were significantly increased by localizing and concentrating the reactants and lowering the energy barrier (Figure 6). [9]
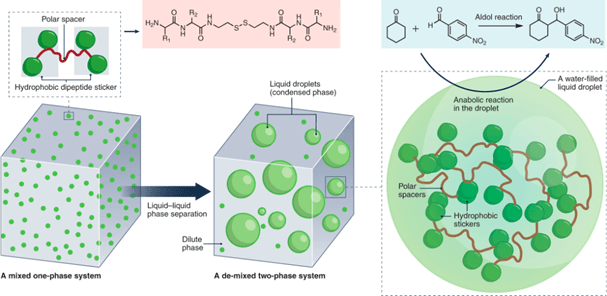
In summary, peptides are the ideal building blocks for creating biomimetic protocells. Increasing the sticker hydrophobicity, β-sheet density, charge density, and total length, or decreasing the polar spacer length tends to lead initially to coacervation, but upon further increase to the formation of solid-like aggregates (Figure 7). The rising discovered peptide-based coacervates make design principles better summarized.
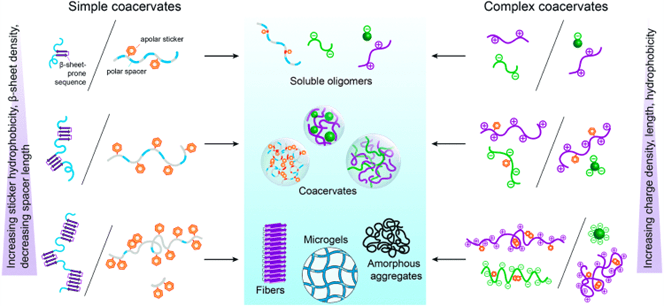
References
- Mehta S, Zhang J. Liquid-liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer 2022, 22(4):239-252.
- Abbas M, Lipiński WP, Wang J, Spruijt E . Peptide-based coacervates as biomimetic protocells. Chem. Soc. Rev. 2021, 50(6):3690-3705.
- Koga S, Williams DS, Perriman AW, Mann S. Peptide-nucleotide microdroplets as a step towards a membrane-free protocell model. Nat. Chem. 2011, 3(9):720-724.
- Saha B, Chatterjee A, Reja A, Das D. Condensates of short peptides and ATP for the temporal regulation of cytochrome c activity. Chem. Commun. (Camb). 2019, 55(94):14194-14197.
- Perry SL, Leon L, Hoffmann KQ, Kade MJ, Priftis D, Black KA, Wong D, Klein RA, Pierce CF 3rd, Margossian KO, Whitmer JK, Qin J, de Pablo JJ, Tirrell M. Chirality-selected phase behaviour in ionic polypeptide complexes. Nat. Commun. 2015, 14;6:6052.
- Li X, Zheng T, Liu X, Du Z, Xie X, Li B, Wu L, Li W. Coassembly of Short Peptide and Polyoxometalate into Complex Coacervate Adapted for pH and Metal Ion-Triggered Underwater Adhesion. Langmuir 2019, 35(14):4995-5003.
- Wei W, Petrone L, Tan Y, Cai H, Israelachvili JN, Miserez A, Waite JH. An Underwater Surface-Drying Peptide Inspired by a Mussel Adhesive Protein. Adv. Funct. Mater. 2016, 26(20):3496-3507.
- Gabryelczyk B, Cai H, Shi X, Sun Y, Swinkels PJM, Salentinig S, Pervushin K, Miserez A. Hydrogen bond guidance and aromatic stacking drive liquid-liquid phase separation of intrinsically disordered histidine-rich peptides. Nat. Commun. 2019, 10(1):5465.
- Abbas M, Lipiński WP, Nakashima KK, Huck WTS, Spruijt E. A short peptide synthon for liquid-liquid phase separation. Nat. Chem. 2021, 13(11):1046-1054.
Clip Chemistry: Diverse (Bio)(macro)molecular and Material Function through Breaking Covalent Bonds
YUAN Dingdong, 27 Oct 2022
The definition of click reaction is “The [click] reaction must be modular, wide in scope, give very high yields, generate only inoffensive byproducts that can be removed by nonchromatographic methods, and be stereospecific (but not necessarily enantioselective). The required process characteristics include simple reaction conditions (ideally, the process should be insensitive to oxygen and water), readily available starting materials and reagents, the use of no solvent or a solvent that is benign (such as water) or easily removed, and simple product isolation.” So, the author borrows this definition and gives the definition of clip reaction “Covalent bond breaking reaction that is modular and wide in scope. It must give very high yields and generate inoffensive byproducts, ideally in a stereo- and regiospecific manner. The process should involve simple reaction conditions and readily available starting materials and reagents.” Here, they give some examples of clip reactions in biomolecular application.
The first type is the Stoichiometric Clip Reaction; stoichiometric clip reactions require the stoichiometric addition of another reagent to trigger bond cleavage. For example, the disulfide reduction reaction. The process is often carried out in the laboratory using a phosphine-reducing agent, as shown in Figure 2a, and in chemical biology, we always used DTT or TCEP as the reductive reagents to reduce their disulfide bond in proteins to break their structure, such as the antibodies, also we use this method to free the cysteine for the next reactions. Also, the silyl ether−fluoride cleavage is another powerful clip reaction, which is most notably known for its role in the protection/ deprotection of alcohols; this method is widely used in organic synthesis; nowadays, this method is also used as the protect part for some biological application such as the drug release (Figure 2b). While the inverse electron demand Diels−Alder reaction of tetrazines and alkenes has been studied since at least the late 1950s, utilized in elegant total syntheses, and more recently popularized as an exceptional click reaction, its development as a clip reaction was only recently reported by Robillard and coworkers. Here, the clip reaction is triggered by a click reaction using modified tetrazines and trans-cyclooctenes and is driven by both dinitrogen extrusion (immediately following the cycloaddition step) and generation of aromaticity (formation of the 1,2-diazine). While these reagents are highly reactive when combined, they are quite stable on their own, giving this system many of the best characteristics of click and clip reactions (Figure 2a and 2c).
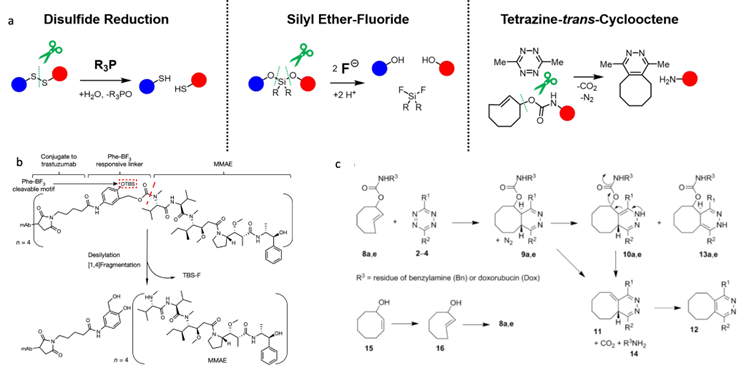
The second type is the Catalytic Clip Reaction; catalytic clip reactions require the presence of an added catalyst to induce bond cleavage. While there are many notable examples, we focus here on hydrolysis (e.g., hydrolysis of acetals, as shown in Figure 3a), palladium-catalyzed dealkylation, and peptide cleavage by proteases (Figure 3a). Palladium-catalyzed deallylation is an example of a catalytic clip reaction that has origins in synthetic organic chemistry but has found powerful recent uses in other fields. Allyl carbamates and carbonates are quite stable in the presence of water. By contrast, upon addition of a palladium catalyst, alkene-coordination activates the carbamate to nucleophilic attack and subsequent cleavage. Following the pioneering work of Tsuji and Trost on palladium-catalyzed allylic alkylation (i.e., the Tsuji−Trost reaction shown in Figure 3b), palladium-catalyzed deprotections of a variety of allylic functional groups quickly emerged, and now such reactions are employed to selectively activate imaging agents and prodrugs in biological systems, such as using Ru metal to achieve the drug decage through antibody-catalyst conjugate.
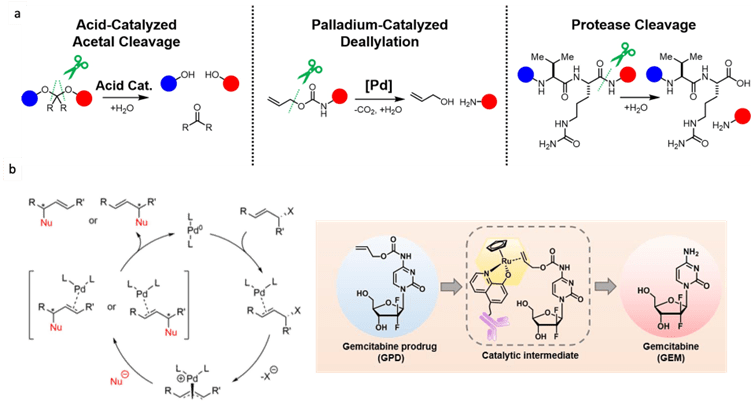
In summary, taking inspiration from the seminal work of Sharpless, Finn, and Kolb outlining the click chemistry concept, they have provided a summary of exceptionally efficient “clip chemistry” processes. The examples given in this review highlight the many opportunities where clip reactions have been used to provide new functions in diverse applications ranging from materials synthesis and characterization to synthetic biology and tissue engineering.
References
- Shieh, P., Hill, M. R., Zhang, W., Kristufek, S. L., & Johnson, J. A. (2021). Clip Chemistry: Diverse (bio)(macro)molecular and material function through breaking covalent bonds. Chemical Reviews, 121(12), 7059–7121.
- Wang, Q., Wang, Y., Ding, J., Wang, C., Zhou, X., Gao, W., Huang, H., Shao, F., & Liu, Z. (2020). A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature, 579(7799), 421–426.
- Zhao, Z., Tao, X., Xie, Y., Lai, Q., Lin, W., Lu, K., Wang, J., Xia, W., & Mao, Z. W. (2022). In situ prodrug activation by an affibody‐ruthenium catalyst hybrid for her2‐targeted chemotherapy. Angewandte Chemie International Edition, 61(26).
Hydrogel in medical biotechnology
HAN Yongxu, 29 Sep 2022
Hydrogels are typically defined as interconnected 3D molecular networks containing a high density of functional groups that enable the absorption and retention of large amounts of water. A wide variety of materials can be used to produce hydrogel networks. These materials can be broadly classified as having either synthetic or naturally occurring components. Synthetic building blocks, including polyacrylic acid, polyethylene glycol and so on. Meanwhile, natural components, including alginate, chitosan, and nucleic acids, in addition to proteins such as collagen, keratin and gelatin that naturally form hydrogels often possess increased biocompatibility and biological functionality when compared to synthetic hydrogels.
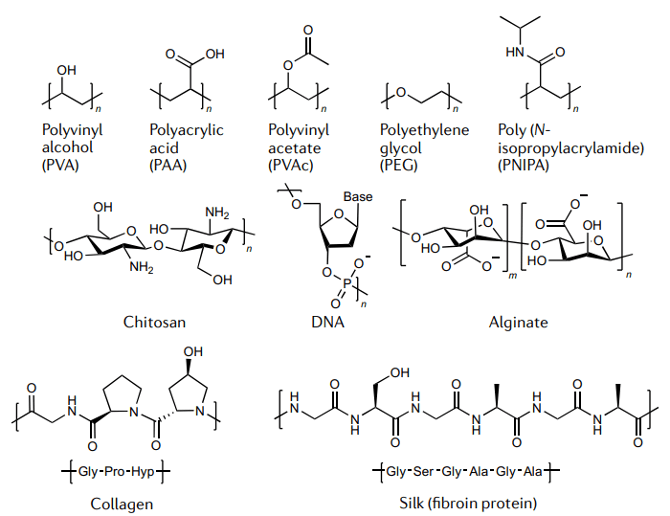
Hydrogel in biomedical exploration like drug delivery, tissue engineering, cell delivery, building scaffolds has become increasingly studied. For example, like Figure 2, the author reported a family of synthetic hydrogels that promote extensive organoid morphogenesis through dynamic rearrangements mediated by reversible hydrogen bonding. These tunable matrices are stress relaxing and thus promote efficient crypt budding in intestinal stem-cell epithelia through increased symmetry breaking and Paneth cell formation dependent on yes-associated protein. As such, these well-defined gels provide promising versatile matrices for fostering elaborate in vitro morphogenesis.
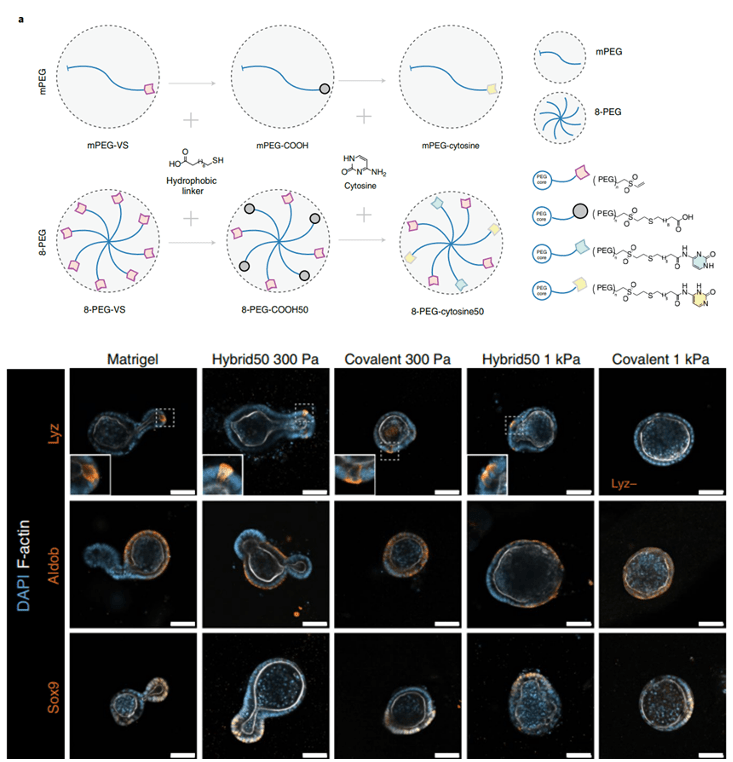
Another report shown in Figure 3, demonstrated that silver nanoparticle (AgNP)-based hydrogel enhances immunotherapy via the modulation of oral microbiota. The authors found a genus of bacteria (Peptostreptococcus) that could activate the immune system and improve the prognosis of patients. They had shown that in mice with subcutaneous or orthotopic murine OSCC tumours, combination therapy with the two components (nanoparticle-incorporating hydrogel and exogenous P. anaerobius) synergized with checkpoint inhibition with programmed death-1.
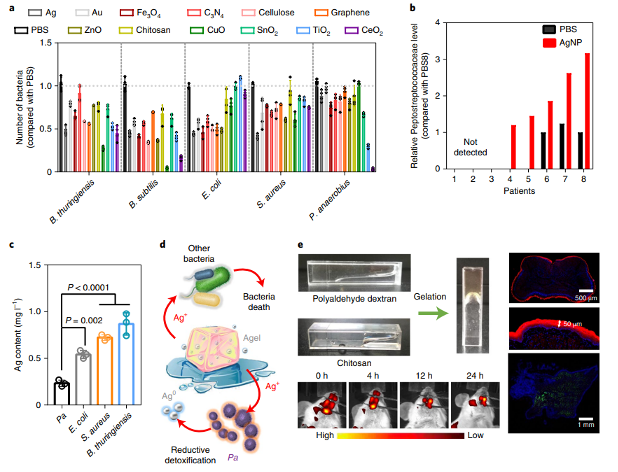
It is worthy mention that there is a simple and efficient method of CAR-T delivery with hydrogel: CAR-T cells and cytokines are added to a specially formulated hydrogel, which provides a temporary, immune cell-activated in vivo environment, and then pumped activated CAR-T cells to attack the tumor tissue. More importantly, this delivery method has the potential to treat distant tumors, which opens new doors for CAR-T therapy in solid tumors.
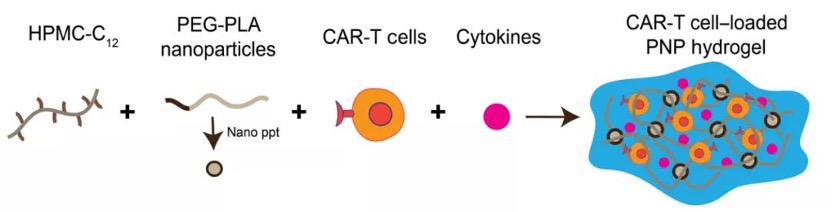
Lastly, the use of HMPs for biofabrication is still in its infancy, there are opportunities at the intersection of research in biomaterials, the microbiota and host diseases with hydrogel from the joint efforts of scientists.
References
- Allen M E, Hindley J W, Baxani D K, et al. Hydrogels as functional components in artificial cell systems. Nature Reviews Chemistry, 2022: 1-17.
- Chrisnandy A, Blondel D, Rezakhani S, et al. Synthetic dynamic hydrogels promote degradation-independent in vitro organogenesis. Nature Materials, 2022, 21(4): 479-487.
- Zheng D W, Deng W W, Song W F, et al. Biomaterial-mediated modulation of oral microbiota synergizes with PD-1 blockade in mice with oral squamous cell carcinoma. Nature Biomedical Engineering, 2022, 6(1): 32-43.
- Grosskopf A K, Labanieh L, Klysz D D, et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors Science advances, 2022, 8(14): eabn8264.
A Brief introduction to electron microscope
JIANG Hao, 19 May 2022
1. The limit of traditional optical microscope
Traditionally, scientists use optical microscope to observe microstructure of biological tissue and cells. But optical microscope have a limit in resolution. The limitation is caused by Rayleigh Criterion. The angular resolution of an optical system can be estimated (from the diameter of the aperture and the wavelength of the light) by the Rayleigh criterion defined by Lord Rayleigh: two point sources are regarded as just resolved when the principal diffraction maximum (center) of the Airy disk of one image coincides with the first minimum of the Airy disk of the other, as shown in Figure. 1.

2. Transmission electron microscope
To reach higher resolution, scientists turn to use electrons to observe the micro world. 1933, Ruska built the first electron microscope that exceeded the resolution attainable with an optical microscope. An electron microscope is a microscope that uses a beam of accelerated electrons as a source of illumination. Electron microscopes use shaped magnetic fields to form electron optical lens systems that are analogous to the glass lenses of an optical light microscope.

The original form of the electron microscope, the transmission electron microscope (TEM), uses a high voltage electron beam to illuminate the specimen and create an image. The electron beam is produced by an electron gun, commonly fitted with a tungsten filament cathode as the electron source. The electron beam is accelerated by an anode, focused by electrostatic and electromagnetic lenses, and transmitted through the specimen that is in part transparent to electrons and in part scatters them out of the beam. When it emerges from the specimen, the electron beam carries information about the structure of the specimen that is magnified by the objective lens system of the microscope. The spatial variation in this information (the “image”) may be viewed by projecting the magnified electron image onto a fluorescent viewing screen, a photographic film or the sensor of a digital camera.
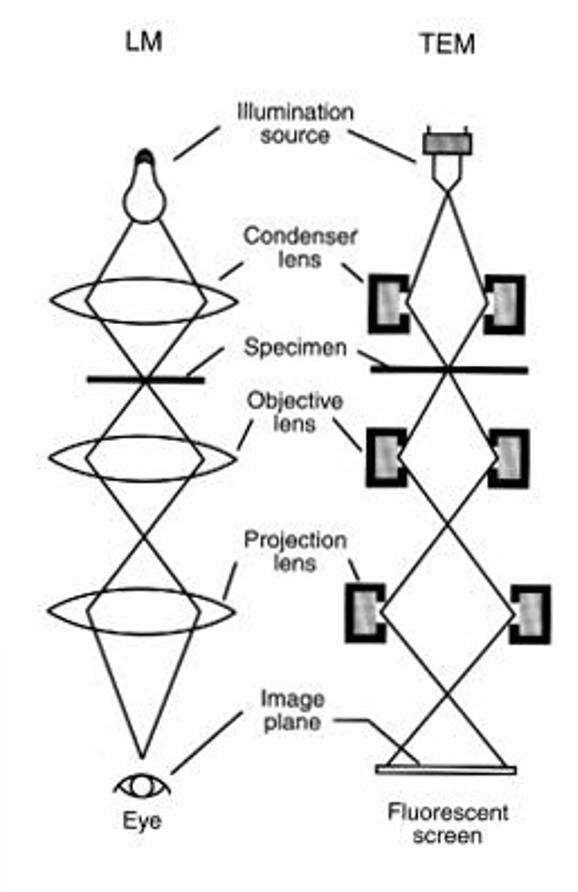
One major disadvantage of the transmission electron microscope is the need for extremely thin sections of the specimens, typically about 100 nanometers. Creating these thin sections for biological and materials specimens is technically very challenging. Semiconductor thin sections can be made using a focused ion beam. Biological tissue specimens are chemically fixed, dehydrated and embedded in a polymer resin to stabilize them sufficiently to allow ultrathin sectioning. Sections of biological specimens, organic polymers, and similar materials may require staining with heavy atom labels in order to achieve the required image contrast.
3. Scanning electron microscope
The SEM produces images by probing the specimen with a focused electron beam that is scanned across a rectangular area of the specimen (raster scanning). When the electron beam interacts with the specimen, it loses energy by a variety of mechanisms. The lost energy is converted into alternative forms which provide signals carrying information about the properties of the specimen surface. The image displayed by an SEM maps the varying intensity of any of these signals into the image in a position corresponding to the position of the beam on the specimen when the signal was generated.

4. Cryogenic electron microscopy
Cryogenic electron microscopy (cryo-EM) is a cryomicroscopy technique applied on samples cooled to cryogenic temperatures. For biological specimens, the structure is preserved by embedding in an environment of vitreous water. An aqueous sample solution is applied to a grid-mesh and plunge-frozen in liquid ethane or a mixture of liquid ethane and propane. While development of the technique began in the 1970s, recent advances in detector technology and software algorithms have allowed for the determination of biomolecular structures at near-atomic resolution. This has attracted wide attention to the approach as an alternative to X-ray crystallography or NMR spectroscopy for macromolecular structure determination without the need for crystallization.

Advancements in direct electron detectors and automation of sample production has led to an increase in use in biological fields, making Cryo-EM a potential rival to X-ray crystallography. The resolution of X-ray crystallography is limited by crystal purity, and creating these samples is very time-consuming, taking up to months or even years. Also, some proteins are hard to crystallize. Although sample preparation for Cryo-EM is still laborious, it does not have these issues as it observes the sample in its “native state”. The median resolution achieved by X-ray crystallography (as of May 19, 2019) on the Protein Data Bank is 2.05 Å. As of 2020, the majority of the protein structures determined by Cryo-EM are at a lower resolution of 3–4 Å. However, the best Cryo-EM resolutions are approaching 1.5 Å, making it a fair competitor in resolution in some cases.
References
- History of electron microscopy, 1931–2000. Authors.library.caltech.edu (2002-12-10). Retrieved on 2017-04-29.
- Rudenberg, H. Gunther; Rudenberg, Paul G. (2010). “Origin and Background of the Invention of the Electron Microscope”. Advances in Imaging and Electron Physics. Vol. 160. pp. 207–286. doi:10.1016/S1076-5670(10)60006-7. ISBN 978-0-12-381017-5.
- Ruska, Ernst (1986). “Ernst Ruska Autobiography”. Nobel Foundation. Retrieved 2010-01-31.
- Tivol, William F.; Briegel, Ariane; Jensen, Grant J. (October 2008). “An Improved Cryogen for Plunge Freezing”. Microscopy and Microanalysis. 14 (5): 375–379. Bibcode:2008 MiMic. 14.375T. doi:10.1017/S1431927608080781. ISSN 1431-9276. PMC 3058946. PMID 18793481.
- Cressey D, Callaway E (October 2017). “Cryo-electron microscopy wins chemistry Nobel”. Nature. 550 (7675): 167. Bibcode:2017 Nature. 550.167C. doi:10.1038/nature.2017.22738. PMID 29022937.
- Yip, Ka Man; Fischer, Niels; Paknia, Elham; Chari, Ashwin; Stark, Holger (2020). “Atomic-resolution protein structure determination by cryo-EM”. Nature. 587 (7832): 157–161. Bibcode:2020Nature. 587.157Y. doi:10.1038/s41586-020-2833-4. PMID 33087927.
Sulfonamide antibiotics and cephalosporins
ZHANG Yu, 12 May 2022
1. Sulfonamide antibiotics
Sulfonamide drugs were the first broadly used antibiotics and paved the way for the antibiotic revolution in medicine. Sulfonamide is a functional group act as competitive inhibitors of the enzyme dihydropteroate synthase (DHPS) which is an enzyme involved in folate synthesis. Folate is vital to the growth and development of the livings. Compared with the bacteria, human can acquire folate though the diet. Therefore, cutting of the folate supply by using the sulfonamide antibiotics can help us inhibit bacterial infection specifically without hurting ourselves. However, as a competitive inhibitor, the sulfonamide antibiotics can only inhibit the growth and multiplication of bacteria, but not kill them.[1]
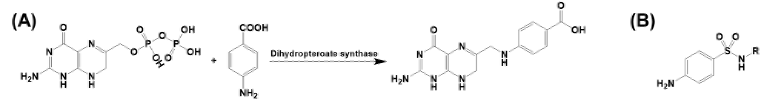
Prontosil, as Bayer named the new drug, was the first medicine ever discovered that could effectively treat a range of bacterial infections inside the body. It had a strong protective action against infections caused by streptococci, including blood infections, childbed fever, and erysipelas, and a lesser effect on infections caused by other cocci. However, it had no effect at all in the test tube, exerting its antibacterial action only in live animals. Later, Daniel Bovet, Federico Nitti, and Jacques and Thérèse Tréfouël found that the drug was metabolized into two parts inside the body, releasing a smaller, colorless, active compound called sulfonamide. [3]
The discovery of Prontosil paved the road for the development of sulfonamide antibiotics and filled the blank of the bacteria infection treatment before Penicillin. With the discovery of Penicillin, the sulfonamide antibiotics slowly faded out from the antibiotic market because of more side effects. However, sulfonamide drugs still work as diuretics or COX-2 inhibitors in pharmaceutical field.
2. Cephalosporins
The cephalosporins are a class of β-lactam antibiotics originally derived from the fungus Acremonium, which was previously known as Cephalosporium. The aerobic mold which yielded cephalosporin C was found in the sea near a sewage outfall in Su Siccu, by Cagliari harbour in Sardinia, by the Italian pharmacologist Giuseppe Brotzu in July 1945, 17 years later than the discovery of Penicillin.[4]

Different with the sulfonamide antibiotics, Cephalosporins, like other β-lactam antibiotics, disrupt the synthesis of the peptidoglycan layer forming the bacterial cell walls. The peptidoglycan layer is important for cell wall structural integrity. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by penicillin-binding proteins (PBPs). PBPs bind to the end of muropeptides to crosslink the peptidoglycan. Beta-lactam antibiotics mimic the ending D-Ala-D-Ala site, thereby irreversibly inhibiting PBP crosslinking of peptidoglycan.[5]
Unlike penicillin, only about 10% of patients with allergic hypersensitivity to penicillin or carbapenems also have cross-reactivity with cephalosporins. This feature makes it a safe and plausible drug in clinic and to slowly take place of penicillin. However, several cephalosporins are associated with a disulfiram-like reaction with ethanol including latamoxef, cefmenoxime, cefoperazone, cefamandole, cefmetazole and cefotetan. Remember: do not drink after using cephalosporins!
References
- Henry RJ (1943). “The Mode of Action of Sulfonamides”. Bacteriological Reviews. 7 (4): 175–262.
- Otten H (1986). “Domagk and the development of the sulphonamides”. Journal of Antimicrobial Chemotherapy. 17 (6): 689–696.
- “History of medicine”. Encyclopædia Britannica. Retrieved 17 January 2014.
- Tilli Tansey; Lois Reynolds, eds. (2000). Post Penicillin Antibiotics: From acceptance to resistance? Wellcome Witnesses to Twentieth Century Medicine. History of Modern Biomedicine Research Group.
- Tipper, D J; Strominger, J L (1965). “Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine”. Proceedings of the National Academy of Sciences of the United States of America. 54 (4): 1133–1141.
GEFs play an important role in the control of small G proteins
QIU Jiaming, 5 May 2022
1. Rho superfamily
Rho GTPases are molecular switches that control a wide variety of signal transduction pathways inside cells. The Ras superfamily consists of more than 150 family members and can be divided into five categories, including five conserved subfamilies – Ras, Rho, Ran, Rab, and Arf(see Figure 1) – each with its own characteristics in terms of sequence, structural similarity, and cellular function. The Ras subfamily contains 36 members responsible for regulating signaling pathways involved in cell proliferation and for regulating signaling pathways involved in cell proliferation, morphology, differentiation, and cellular functions. The largest subfamily of the Ras superfamily is Rab, which consists of more than 60 members involved in vesicle and protein transport in cells. 30 members of the Arf family act as regulators of intracellular transport. The Ras subfamily consists of 36 members and is responsible for regulating signaling pathways involved in cell proliferation. The 20 members of the Rho subfamily are responsible for the regulation of signaling pathways involved in cell proliferation, transport and differentiation, and cell survival. The 20 members of the Rho subfamily are key regulators of actin organization, gene expression and cell cycle progression. The largest one subfamily, Rab, consists of more than 60 members and is involved in vesicle and protein transport. involved in vesicle and protein transport. There are 30 members of the Arf protein members that are also regulators of intracellular transport.Ran subfamily. The Ran subfamily has only one protein, which is the most abundant small GTPase in the cell. It is the most abundant small GTPase in the cell and is involved in nuclear transport.
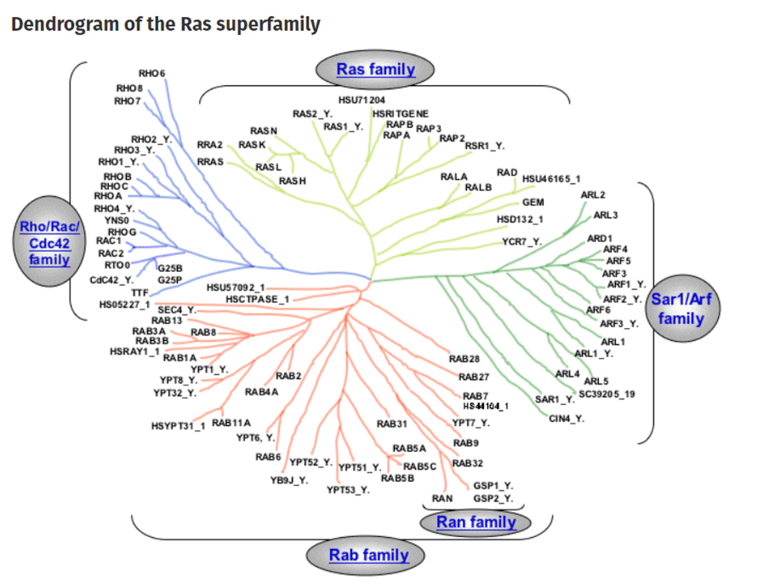
2. GEFs and GAPs
Guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) regulate the activity of small guanine nucleotide-binding (G) proteins for the purpose of regulating cellular functions (See Figure2). In general, GEFs regulate signaling by catalyzing the exchange of G protein-bound GDP to GTP, whereas GAPs terminate signaling by inducing GTP hydrolysis. Recent evidence suggests that these proteins may be potential therapeutic targets for the development of drugs to treat a variety of diseases, including anticancer antibacterial. GEFs contain a variety of species, all of which activate Cdc42 by mediating the exchange of GDP with GTP in Cdc42. during activation, when receiving external signals, GEFs such as Dbl bind to inactive Cdc42, forming a Cdc42-GEF binary complex, causing GDP binding to fall off from small G proteins, followed by the release of GTP from Cdc42 due to the cell membrane when GTP can be more than ten times that of GDP. GEF binds with low affinity to active Cdc42 and is therefore released from Cdc42-GTP. Active Cdc42 interacts with downstream effectors such as N-WASP to produce biological functions such as actin polymerization. GTPase activating proteins or GAPs promote GTP hydrolysis of Cdc42 and return Cdc42 to its basal inactive state
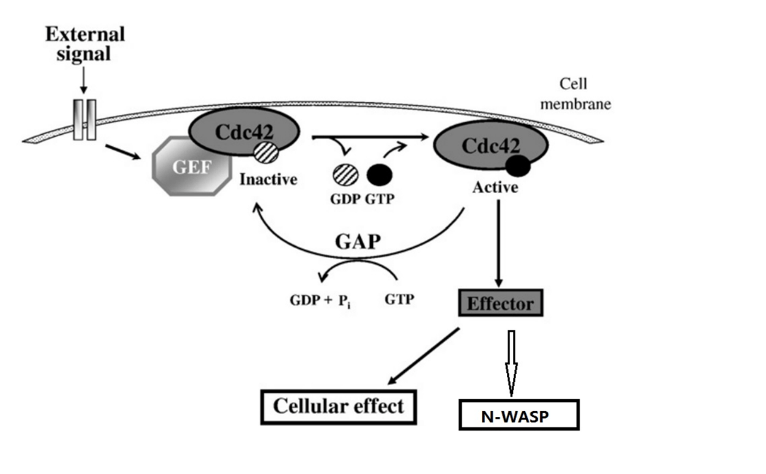
3. Cdc42
Human Cdc42 is a small GTPase of the Rho family that plays a very important role in the cellular life cycle, including the regulation and control of intracellular signaling pathways such as cell morphology, cell migration, cell polarization, and cell cycle progression. Rho GTPases are key factors in regulating intracellular actin cytoskeleton assembly and reorganization, which can regulate cell function by regulating is cell adhesion and migration, as well as being major players in pathogen infestation of host cells such as EPEC. Activation of Cdc42 requires regulation by members such as GEFs to undergo conformational changes, and from cdc42GDP to cdc42GTP transformation, which initiates the reorganization of actin and regulates the adhesion, migration and invasion of cells as well as pathogenic microorganisms. Thus, the regulation of cdc42 affects numerous physiological functions of cells and has become a hot topic of research.
References
- Takai et al. Small GTP-Binding Proteins. Physiol. 2001
- Jeffrey R et al. Proceedings of the National Academy of Sciences, 2001
- Soniya Sinha, Wannian Yang. Cellular signalling. 2008
Emerging New Concepts of Degrader Technologies
YUAN Dingdong, 28 April 2022
Human genetic studies have shown that many novel target proteins cause disease by acquiring functional toxicity. Traditional drug development strategies need to occupy binding sites to inhibit the functional activity of target proteins, which makes disease-related scaffold proteins, transcription factors, and other non-enzymatic proteins “undruggable”. These “undruggable” disease-causing proteins can be corrected or degraded by enhancing protein quality control systems.
Selective degradation of pathogenic protein levels is a more effective strategy. Nucleic acid-based RNA- or DNA-targeting agents for gene silencing are one approach, while gene therapy is promising, it still faces some key challenges such as being difficult to deliver and prohibitively expensive. Therefore, inducing degradation of target proteins is more feasible and can meet clinical needs (Fig. 1).1
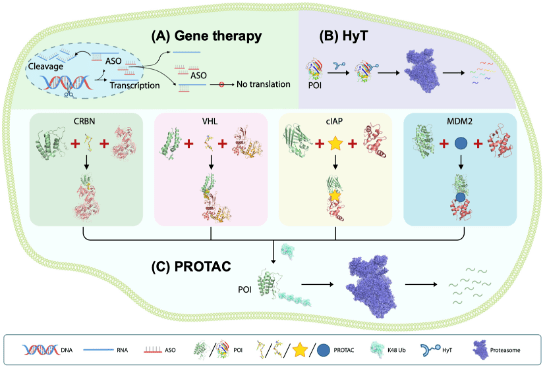
- 2. Total synthesis of quinine
Small molecules are able to induce selective degradation of the protein of interest (POI) by adding tags recognized by the degradation machinery. In 2011-2012, Crews and colleagues reported POI degradation induced by small molecule hydrophobic tags (HyT). HyT is achieved by a molecule composed of a hydrophobic segment and a ligand that binds to POI. Thus, HyT can add hydrophobic fragments to the POI, resulting in structural changes of the target protein and subsequent proteolysis (Fig. 1).1
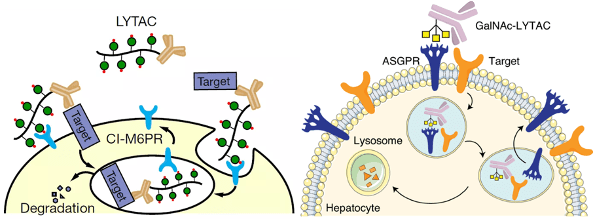
PROTAC
At present, the main method of small molecule-induced protein degradation is the protein degradation targeted chimera (PROTAC) technology. The PROTAC molecule increases the ubiquitination efficiency of the target protein by shortening the distance between the target protein and E3 ligase, thereby inducing the proteasome system to degrade the target protein. However, PROTAC has certain limitations. For example, this method relies on a specific E3 ligase, which limits its application in certain cells. Its molecular weight is generally too large, and it cannot target the secret proteins and membrane proteins (Fig. 1).1
LYTAC
LYTAC (Lysosome Targeted Chimera) technology is a method that utilizes the endosome/lysosome pathway to degrade target proteins. PROTAC technology mainly targets intracellular proteins, while LYTAC can act on membrane proteins and extracellular proteins that are resistant to PROTAC (such as EGFR). LYTAC molecules consist of antibodies against a specific target protein and are covalently linked to mannose 6-phosphate (M6P). Antibodies are recognized by endogenous systems, and their bound target proteins are transported to lysosomes for degradation. The advantage of LYTAC technology is that it utilizes the endogenous degradation pathway to degrade extracellular proteins and membrane proteins. The main disadvantage is that the molecular weight is large, and the antibody or polypeptide in the molecule may induce an immune response (Fig. 2).2

In March 2021, they published a new work in the journal Nature Chemical Biology, revealing a LYTAC technology that utilizes the asialoglycoprotein receptor (ASGPR). Since ASGPR is a liver-specific lysosomal targeting receptor, LYTAC technology utilizing ASGPR is able to degrade extracellular proteins in a cell-type-restricted manner. Specifically, the study conjugated the binder triantenerrary N-acetylgalactosamine (tri-GalNAc) of ASGPR to the target protein through a linker. Based on these findings, the researchers believe that GalNAc-LYTAC represents a cell-type-restricted lysosomal protein degradation pathway that further paves the way for the clinical translation of LYTAC technology (Fig. 2).3
AUTAC
Autophagy-targeted chimeras (AUTACs) have a similar design to PROTACs and are well suited to degrade cytoplasmic target proteins resistant to PROTAC molecules. POIs are first phagocytosed to form autophagosomes, and then autophagy receptors such as SQSTM1/p62 recognize Lys63 (K63) polyubiquitinated POIs and transfer them to autophagosomes for degradation. Both AUTAC and PROTAC molecules act through ubiquitination, but AUTAC molecules induce target degradation by triggering K63 polyubiquitination. The study found that AUTAC molecules can degrade target proteins and damaged organelles such as mitochondria (Fig. 3).4
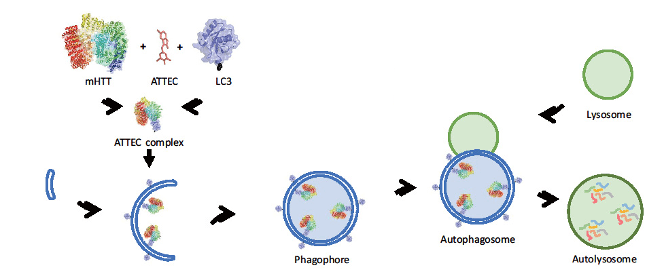
ATTEC
Autophagosome-binding compounds (ATTECs) are a more direct approach to utilize autophagy to degrade target proteins. Unlike PROTAC and AUTAC, ATTEC molecules are not associated with ubiquitination. ATTEC molecules can directly bind the target protein to LC3 and promote the phagocytosis of the target protein by autophagosomes. mHTT (mutated mHTT) is the main cause of Huntington’s disease (HD), and this protein has repeat-expanded polyglutamine (polyQ). Studies have shown that autophagosome-conjugated compounds can degrade mHTT in cells or animal models and attenuate HD-related phenotypes. In vitro experiments showed that ATTEC molecules can interact specifically with polyQ to degrade mHTT without affecting the level of wtHTT selectively. ATTEC molecules are also able to degrade other disease-causing polyQ proteins. By directly interacting with the autophagosome protein LC3 and bypassing the ubiquitination process, ATTEC molecules have great potential to degrade DNA/RNA molecules, damaged organelles, and other non-protein cargoes recognized by autophagy. Whether ATTEC molecules affect overall autophagic activity remains to be investigated. Compared with the molecular weights of PROTAC, LYTAC, and AUTAC, the molecular weight of ATTEC targeting mHTT is very small, which may have better drug properties (Fig. 4).5
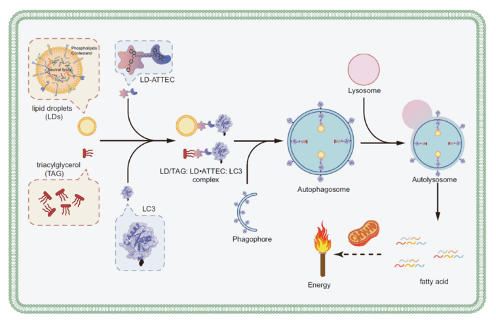
The project team designed ATTEC compounds (LD-ATTEC) with lipid droplets, the intracellular fat-storing organelles, as degradation targets. Excessive accumulation of lipid droplets may be associated with various diseases, such as obesity, non-alcoholic fatty liver disease, and neurodegeneration. Therefore, the degradation of lipid droplets may have beneficial effects on related diseases, and LD-ATTEC can also be used as a tool compound to study the causal association of lipid droplets with these diseases (Fig. 5).6
The lysosomal pathway can theoretically degrade various biological macromolecules and even organelles. On the other hand, lysosomes can also degrade proteins that were originally degraded by the proteasome. Therefore, lysosome-targeted degradation technology may theoretically degrade various pathogenic proteins, protein aggregates, DNA/RNA, organelles, pathogens, lipids, peroxisomes, and other disease-related substances.
LYTAC, AUTAC, and ATTEC, these lysosome targeting technologies, have different principles. LYTAC mainly utilizes the endosome-lysosome pathway, which is suitable for extracellular and cell membrane proteins. AUTAC is based on selective autophagy mediated by K63 ubiquitination and can act on intracellular proteins that K63 can ubiquitinate. ATTEC directly binds the target to be degraded to the autophagosome, so the target range that can be degraded is not limited to proteins.
References
- Ding, Y., Fei, Y., & Lu, B. (2020). Emerging New Concepts of Degrader Technologies. Trends In Pharmacological Sciences, 41(7), 464-474. doi: 10.1016/j.tips.2020.04.005
- Banik, S., & Bertozzi, C. (2020). Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature, 584(7820), 291-297. doi: 10.1038/s41586-020-2545-9
- Roggenbuck, D., Mytilinaiou, M., Reinhold, D., & Conrad, K. (2012). Asialoglycoprotein receptor (ASGPR): a peculiar target of liver-specific autoimmunity. Autoimmunity Highlights, 3(3), 119-125. doi: 10.1007/s13317-012-0041-4
- Takahashi, D., Moriyama, J., Itto-Nakama, K., & Arimoto, H. (2019). AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Molecular Cell, 76(5), 797-810.e10. doi: 10.1016/j.molcel.2019.09.009
- Li, Z., Wang, C., Wang, Z., Ding, C., Ding, Y., Fei, Y., & Lu, B. (2019). Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature, 575(7781), 203-209. doi: 10.1038/s41586-019-1722-1
- Fu, Y., Chen, N., Ding, Y., & Lu, B. (2021). Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Research, 31(9), 965-979. doi: 10.1038/s41422-021-00532-7
Story of Quinine: Structure, History, and Total Synthesis
BAO Yishu, 21 April 2022
1. Discovery of quinine
Tonic water glows faintly in sunlight and this phenomenon is even more apparent under ultraviolet light irradiation. It arises from tonic water’s main ingredient, quinine. The glow of a quinine solution was firstly noticed by the polymath Sir John Herschel.1 A few years later, this phenomenon was called “fluorescence” by the physicist and mathematician Sir George Stokes.
Quinine is an alkaloid extracted from the bark of Cinchona trees indigenous to South American forests, which attracted intense interest worldwide on account of its prophylactic characteristics. Its name is derived from quina-quina and it can be roughly translated as bark of barks. Native South Americans used it to alleviate shivering, fever, and pain.2 Later, they found that quinine also promptly relieved the symptoms of malaria and quinine became well-known around the world for it. Before artemisinin was discovered, quinine was the drug of choice for the treatment of malaria (Figure 1).

- 2. Total synthesis of quinine
The chemical structure of quinine is complex and its total synthesis confounded chemists for over a century since its first isolation from the bark in 1820 by two French pharmacists. In 1856, Sir William Henry Perkin who was only 18 years old, attempted to synthesize quinine by oxidizing N-allyltoluidine. He failed but he generated mauveine which is the first synthetic organic dye to be synthesized on a large scale for industrial and commercial purposes (Figure 2).3 Subsequent attempts by Paul Rabe and Karl Kindler in the early twentieth century. This sequence involved: (1) oxidation of d-quinotoxine with sodium hypobromite to produce N-bromoquinotoxine; (2) base-mediated cyclization to produce quininone; and (3) aluminum powder reduction of quininone to produce quinine and quinidine (Figure 3).4
Regarding the breakthrough progress in the total synthesis of quinine, we must mention an organic chemistry legend, Robert Burns Woodward. Woodward was born in Boston in 1917. In 1933, 16-years-old Woodward enrolled as an undergraduate at Massachusetts Institute of Technology (MIT). In 1936, he received a Bachelor of Science degree. Only one year later, MIT awarded him a doctorate. In 1944, World War II broke out, which made the spread of malaria especially fast, resulting in a short supply of the quinine. At this time, Woodward made an amazing decision to totally synthesize quinine. He and his postdoctoral researcher William von Eggers Doering reported the synthesis of quinine, starting from using 3-hydroxybenzaldehyde, through 55 reactions including condensation, cyclization, alkylation, reduction, acetylation, oxidation, and so on. The final step is the Claisen condensation and finally, the generation of the racemic quinidone after acid treatment (Figure 4).5
In addition to quinine, Woodward and his research group’s achievements in total synthesis are breathtaking: cholesterol, cortisone, lanosterol, strychnine, tetracylines, colchicine, reserpine, lysergic acid and ergonovine, chlorophyll, cephalosporin C, erythromycin, and vitamin B12 (the last, with Albert Eschenmoser). Those are just a few among the herd of his synthetic accomplishments. In 1965, Woodward won the Nobel Prize in Chemistry for his outstanding achievements in organic synthesis.
However, the total synthesis of quinine reported by Woodward and Doering did not address the stereoselective problem. In 2001, Gilbert Stork reported the first entirely stereoselective total synthesis of (−)-quinine (Figure 5).6 The Staudinger reaction involved in the synthetic route proposed by Stork was developed by Bertozzi’s group as a bioorthogonal reaction, the Staudinger coupling reaction in 2000 (Figure 6).7 the Staudinger ligation is a modification of the classical Staudinger reaction in which an electrophilic trap is placed on the triaryl phosphine. In aqueous media, the aza-ylide intermediate rearranges, to produce an amide linkage and the phosphine oxide, and is so named the Staudinger ligation because it ligates two molecules together, whereas, in the classical Staudinger reaction, the two products are not covalently linked after hydrolysis. Staudinger ligation has been used for the labeling of glycans, lipids, DNA, and proteins.
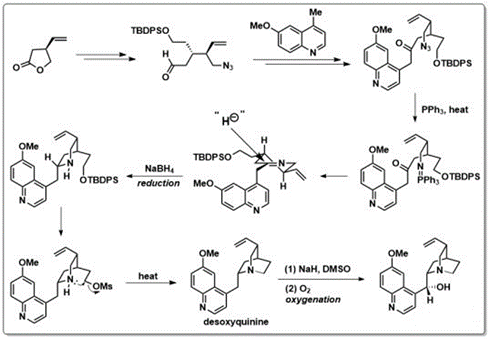
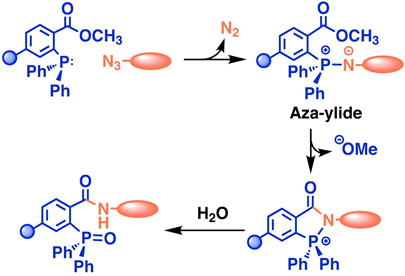
In summary, the total synthesis of quinine is too complicated. Still, the main source of commercial quinine today remains Cinchona bark. Quinine serves to both treat and prevent malaria, but how precisely it exerts its killing effect on the virulent Plasmodium falciparum parasite has so far remained elusive. There is much evidence to indicate that it interferes with the parasite’s haemoglobin degradation pathway. In addition, a recent study used a cellular thermal shift assay coupled with mass spectrometry to identify a protein target, P. falciparum purine nucleoside phosphorylase (PfPNP) (Figure 7).8
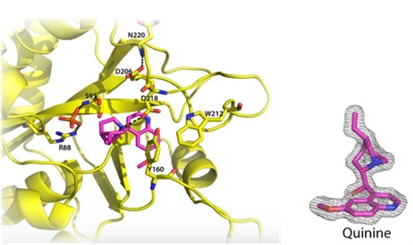
Nowadays, the Cinchona alkaloid is still ubiquitous. It continues to be clinically used as a medicine to treat severe babesiosis, its blue glow serves as a fluorescence standard, and it is a useful ligand in asymmetric organocatalysis. It also remains a key ingredient in tonic water.
References
- Herschel, J. F. W. Phil. Trans. R. Soc. Lond. 1845, 135, 143-145.
- Kaufman, T. S. Rúveda, E. A. Angew. Chem. Int. Ed. 2005, 44, 854-885.
- Perkin, W. H. J. Chem. Soc. Trans. 1896, 69, 596-637.
- Smith, A. C. Williams, R. M. Angew. Chem. Int. Ed. 2008, 47, 1736-1740.
- Woodward, R. B. Doering, W. E. J. Am. Chem. Soc. 1945, 67, 860-874.
- Stork, G. Niu, D. Fujimoto, R. A. Koft, E. R. Balkovec, J. M. Tata, J. R. Dake, G. R. J. Am. Chem. Soc. 2001, 123, 3239-3242.
- Saxon, E. Bertozzi, C. R. Science 2000, 287, 2007-2010.
- Dziekan, J. M. Yu, H. Chen, D. Dai, L. Wirjanata, G. Larsson, A. Prabhu, N. Sobota, R. M. Bozdech, Z. Nordlund, P. Sci. Transl. Med. 2019, 11, eaau3174.
Photobiocatalysis-a new way for transformation
CHEN Hongfei, 14 April 2022
1. The era of antibiotic discovery
Biocatalysis reaction especially enzyme reaction has advantages in high chemical selectivity and good stereoselectivity. However, this kind of reaction always has limited substrates scope. On the other hand, photocatalysis reaction which using visible light as energy source can generate open-shell intermediates for different reactions. But the poor stereoselectivity is one of the drawbacks of this kind of reaction. Recent years, researchers combine the biocatalysis and photocatalysis together for new transformations which based on visible light and enzymes. The photobiocatalysis are divided into three main parts: a) natural photoenzymes; b) new reactivity of cofactor dependent enzymes; c) synergistic combination of photocatalyst and enzymes (Fig.1.).[1]
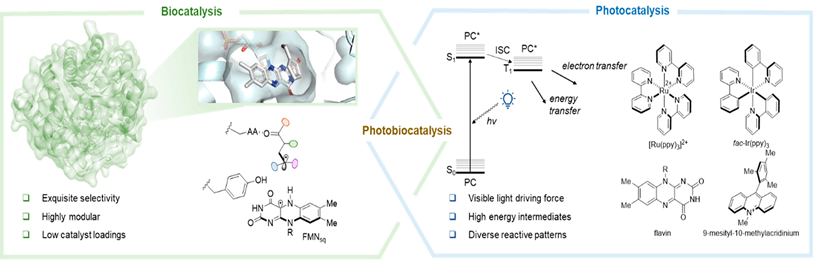
The first kind of photobiocatalysis is natural photoenzyme. A recent example is the discovery and characterization of a fatty acid photodecarboxylase (CvFAP). CvFAP was first reported by Beisson and co-workers in 2017. This enzyme can use FAD and light to produce alkanes and alkenes from long-chain fatty acids (Fig.2.). [2,3]
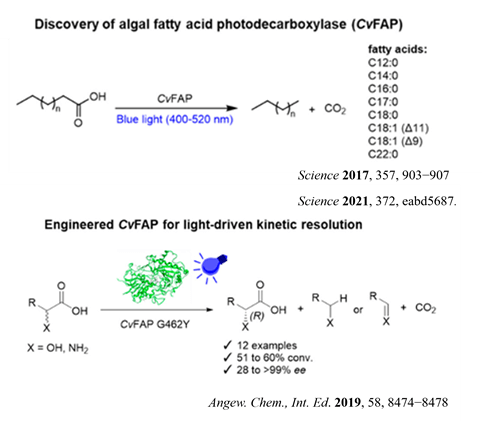
Since the discovery of the newly characterized CvFAP, a variety of photoenzymatic transformations have been disclosed by tuning substrate specificity and function. Very recent studies of engineering CvFAP for a photodecarboxylative deuteration and for a selective photodecarboxylation of trans fatty acids were reported by Wu and co-workers. Given the growing number of applications using this newly discovered CvFAP, it is sure that the discovery of other new photoenzymes will enable the development of new to- nature chemical transformations.[4]
The second kind of photobiocatalysis is new reactivity of cofactor dependent enzymes. The redox active cofactors of these enzymes can form electron donor−acceptor (EDA) complexes with unnatural substrates which can generate radical species under mild conditions through single-electron transfer (SET) events. In 2016, Hyster and co-workers demonstrated the first example of the use of photoexcited enzyme−substrate complexes for achieving non-natural reactivity in enzymes. They discovered NADPH-dependent ketoreductases, which normally catalyze the nonlight-driven reduction of carbonyl functional groups, could catalyze the radical dehalogenation of halolactones through the photoexcitation of a halolactone-NADPH EDA complex. Very recently, they expanded the scope of the reductive cyclization to chiral tertiary amine synthesis by altering the radical acceptor from a tethered C=C double bond to an oxime (Fig.3.). [5,6,7]
The third kind of photobiocatalysis is synergistic combination of photocatalyst and enzymes. Currently, there are three strategies to synergize external photoredox catalysts and enzymes for abiological transformations. In each strategy, the enzyme is used to control selectivity of the reaction, and the external photoredox catalyst can be used for (I) selective single-electron reduction of the enzyme-bound substrate, (II) substrate racemization enabled by the photoinduced SET, or (III) E/Z-isomerization of alkenes via energy transfer. In the last five years, researchers have leveraged the reactivity of photocatalysts and selectivity of enzymes for difficult asymmetric syntheses. It is expected that emerging strategies for dual photocatalysis, and enzyme catalysis can be applied to enzymes without light absorbing cofactors and that the compatibility of photocatalysts with enzyme cofactors could be further explored for abiological transformations. [8,9,10]
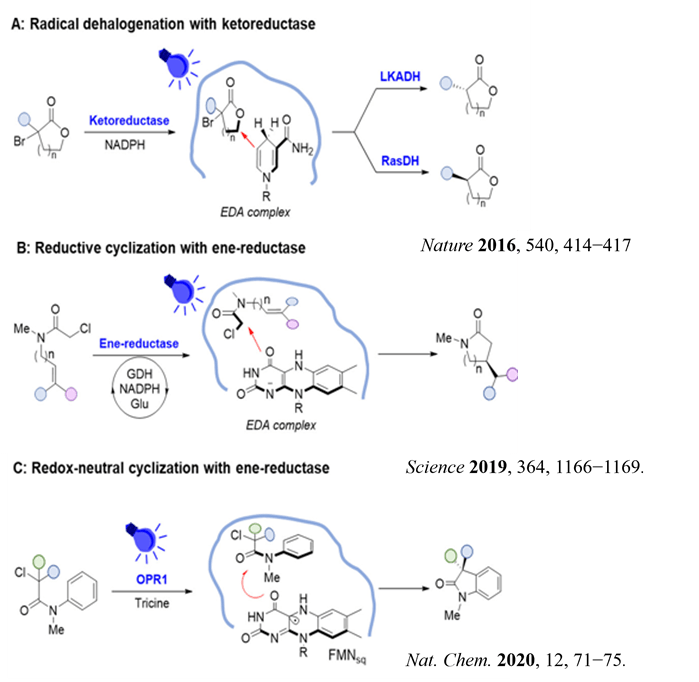
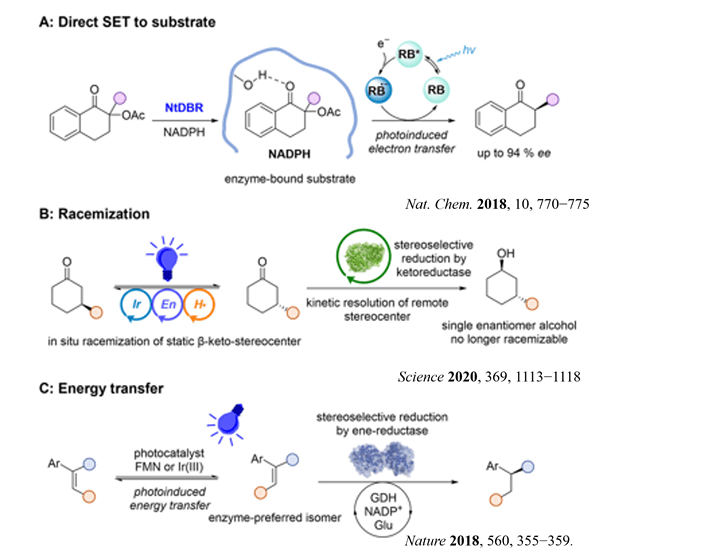
Compared to the booming development of photocatalysis and biocatalysis, the application of photobiocatalysis for the discovery and development of non-natural chemical transformations is still developing at a much slower pace. In the future, more chemical reactions can be discovered and potential interest in exploring the scalability of photobiocatalytic processes for industrial manufacturing.
References
[1] Wesley Harrison, Xiaoqiang Huang, and Huimin Zhao. Photobiocatalysis for Abiological Transformations. Acc. Chem. Res. 2022, 55, 8, 1087–1096
[2] Sorigué, D.; Légeret, B.; Cuiné, S.; Blangy, S.; Moulin, S.; Billon, E.; Richaud, P.; Brugière, S.; Couté, Y.; Nurizzo, D.; Müller, P.; Brettel, K.; Pignol, D.; Arnoux, P.; Li-beisson, Y.; Peltier, G.; Beisson, F. An Algal Photoenzyme Converts Fatty Acids to Hydrocarbons. Science 2017, 357, 903−907
[3] Sorigué, D.; Hadjidemetriou, K.; Müller, P.; Beisson, F. Mechanism and Dynamics of Fatty Acid Photodecarboxylase. Science 2021, 372, eabd5687.
[4] Li, D.; Han, T.; Xue, J.; Xu, W.; Xu, J.; Wu, Q. Engineering Fatty Acid Photodecarboxylase to Enable Highly Selective Decarboxylation of trans Fatty Acids. Angew. Chem., Int. Ed. 2021, 60, 20695−20699.
[5] Emmanuel, M. A.; Greenberg, N. R.; Oblinsky, D. G.; Hyster, T. K. Accessing Non-Natural Reactivity by Irradiating Nicotinamide- Dependent Enzymes with Light. Nature 2016, 540, 414−417.
[6] Biegasiewicz, K. F.; Cooper, S. J.; Gao, X.; Oblinsky, D. G.; Kim, J. H.; Garfinkle, S. E.; Joyce, L. A.; Sandoval, B. A.; Scholes, G. D.; Hyster, T. K. Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science 2019, 364, 1166−1169.
[7] Black, M. J.; Biegasiewicz, K. F.; Meichan, A. J.; Oblinsky, D. G.; Kudisch, B.; Scholes, G. D.; Hyster, T. K. Asymmetric Redox- Neutral Radical Cyclization Catalysed by Flavin-Dependent ‘Ene’- Reductases. Nat. Chem. 2020, 12, 71−75.
[8] Biegasiewicz, K. F.; Cooper, S. J.; Emmanuel, M. A.; Miller, D. C.; Hyster, T. K. Catalytic Promiscuity Enabled by Photoredox Catalysis in Nicotinamide-Dependent Oxidoreductases. Nat. Chem. 2018, 10, 770−775
[9] DeHovitz, J. S.; Loh, Y. Y.; Kautzky, J. A.; Nagao, K.; Meichan, A. J.; Yamauchi, M.; MacMillan, D. W. C.; Hyster, T. K. Static to Inducibly Dynamic Stereocontrol: The Convergent Use of Racemic β-Substituted Ketones. Science 2020, 369, 1113−1118
[10] Litman, Z. C.; Wang, Y.; Zhao, H.; Hartwig, J. F. Cooperative Asymmetric Reactions Combining Photocatalysis and Enzymatic Catalysis. Nature 2018, 560, 355−359.
Computer-aided engineering of enzymatic selectivity
WANG Yue, 7 April 2022
Life has evolved an astonishing array of enzymes that play a wide range of roles in vital biological processes. Intriguingly, enzymes with such remarkable power, selectivity, and sustainability strongly encourage biochemists to supplement or even replace some traditional chemical synthesis routes on a large scale. Although the trend toward biosynthesis of natural products or blockbuster drugs in diverse industries strongly drives the development of enzymes that are capable of catalyzing challenging reactions, even non-natural ones, there is still a mismatch between the inefficiency of enzymes toward non-native substrates and the need for complex chemicals in diverse industries.
The development of credible mechanistic insights requires a high-quality structure in knowledge-based protein engineering. Compared to decades ago, enzyme crystallization has advanced significantly, but is still a much slower process than sequence accumulation. As a result, a variety of computer-based prediction approaches are available that can effectively provide acceptable enzyme structures, which are mainly homology alignments based on templates and conformational sampling strategies without templates (Kuhlman and Bradley, 2019). Meanwhile, machine learning has also been used to explore deeper buried information in structure databases to improve the accuracy of structure prediction, especially in the case of the latest alphafold2 in CASP14 (Service, 2020).
By interacting with the substrate, enzymes perform their catalytic function. As a fundamental tool to predict the optimal binding mode of ligands and enzymes, molecular docking was inspired by “lock-and-key” (Fischer, 1894), “induced-fit” (Koshland, 1958), and “configurational selection” (Kumar et al., 2000) models. These approaches included rigid docking, semi-flexible docking, flexible docking, and the latest CaverDock, which considered substance transport in enzyme channels. The precondition for excellent molecular docking results is the use of high-quality structures, which can be obtained either experimentally or computationally.
Static snapshot structures are often inadequate to analyze dynamic enzyme-catalyzed processes, including substrate binding, chemical bond cleavage, and product release. Dynamic conformational ensembles are continuously sampled using MD simulation, a powerful computational scheme based on classical mechanics (Hollingsworth and Dror, 2018). Additionally, MD simulations of large biosystems are approaching experimentally relevant timescales, raising the possibility of mutually reinforcing experimental and theoretical findings, providing new opportunities in rationalizing and engineering enzymes (Cerutti and Case, 2019; Franz et al., 2020; Huggins et al., 2019; Surpeta et al., 2020). At present, various useful MD methods have been developed, such as MD simulations incorporating enhanced sampling to reveal ligand migration (Rydzewski and Nowak, 2017) and coarse-grained MD simulations to cope with multiscale biological processes (Kmiecik et al., 2016; Souza et al., 2020).

References
Kuhlman, B., Bradley, P., 2019. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 20 (11), 681–697.
Service, R.F, 2020. The game has changed. AI triumphs at solving protein structures. Science.
Fischer, E., 1894. Einfluss der configuration auf die wirkung der enzyme. Berichte der deutschen chemischen Gesellschaft 27 (3), 2985–2993.
Koshland, D.E., 1958. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U. S. A. 44 (2), 98–104.
Kumar, S., Ma, B., Tsai, C.-J., Sinha, N., Nussinov, R., 2000. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci.9 (1), 10–19.
Hollingsworth, S.A., Dror, R.O., 2018. Molecular dynamics simulation for all. Neuron 99(6), 1129–1143.
Cerutti, D.S., Case, D.A., 2019. Molecular dynamics simulations of macromolecular crystals. WIREs Comput. Mol. Sci 9 (4).
Franz, F., Daday, C., Gr¨ater, F., 2020. Advances in molecular simulations of protein mechanical properties and function. Curr. Opin. Struct. Biol. 61, 132–138.
Huggins, D.J., Biggin, P.C., D¨amgen, M.A., Essex, J.W., Harris, S.A., Henchman, R.H., Khalid, S., Kuzmanic, A., Laughton, C.A., Michel, J., Mulholland, A.J., Rosta, E., Sansom, M.S.P., van der Kamp, M.W., 2019. Biomolecular simulations: from dynamics and mechanisms to computational assays of biological activity. WIREs Comput. Mol. Sci 9 (3).
Surpeta, B., Sequeiros-Borja, C.E., Brezovsky, J., 2020. Dynamics, a powerful component of current and future in silico approaches for protein design and engineering. Int. J. Mol. Sci. 21 (8), 2713.
Rydzewski, J., 2020. maze: heterogeneous ligand unbinding along transient protein tunnels. Comput. Phys. Commun 247, 106865.
Kmiecik, S., Gront, D., Kolinski, M., Wieteska, L., Dawid, A.E., Kolinski, A., 2016. Coarsegrained protein models and their applications. Chem. Rev. 116 (14), 7898–7936.
Souza, P.C.T., Thallmair, S., Conflitti, P., Ramírez-Palacios, C., Alessandri, R., Raniolo, S., Limongelli, V., Marrink, S.J., 2020. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 11 (1), 3714.
Antibiotic resistance crisis-the sword of Damocles hanging in the human head
Chunjian Wu, 24 March 2022
1. The era of antibiotic discovery
For most of human history, bacterial pathogens have been a major cause of disease and mortality. In thepre-antibiotic era of the early 1900s, people had no medicines against these common germs and as a result, human suffering was enormous. Death from infection could follow from something as minor as a scratch. But this situation changes after the first antibiotic—penicillin was discovered in 1928 by Scottish scientist Alexander Fleming. Suddenly, the morbidity and mortality sharply were reduced by this“miracle drugs”, which also raises a lot of attention all over the world. Shortly afterward, the golden era of antibiotic launched, and the main classes of antibiotics were discovered in a fairly short period of time (Figure 1).
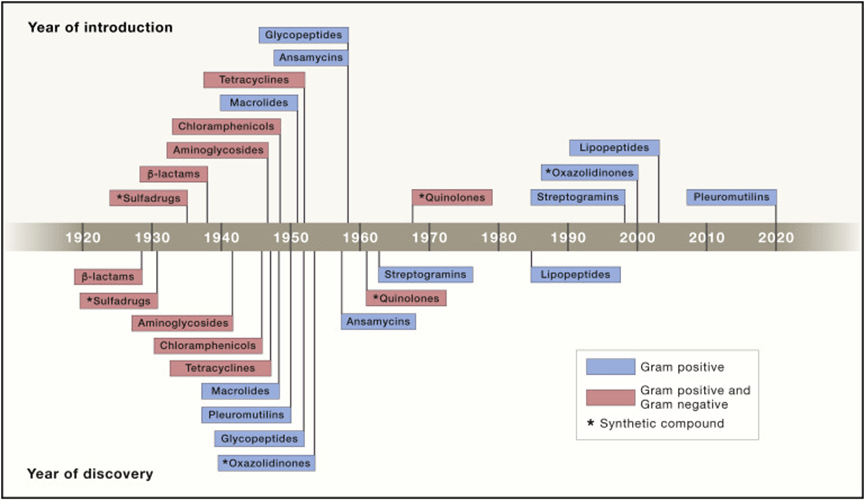
Antibiotics have revolutionized the field of medicine. They have saved millions of lives each year, alleviated pain and suffering, and have even been used prophylactically for the prevention of infectious diseases. What is more, antibiotics have been crucial in the increase in life expectancy in the United States from 47 years in 1900 to 74 years for males and to 80 years for females in the year 2000 (The Journal of antibiotics, 70(5), pp.520-526.). But the discovery of antibiotic resistance appeared as a disturbing storm-cloud in this clear sky.
The crisis of antibiotic resistance
In fact, such a continuous progress in discovery of new antibiotics in the 1940–1980 period was mostly fostered by the necessity of “fighting new antibiotic resistances,” where the new discoveries compensated for the emergence of new resistances in bacterial pathogens. The golden era ended rather abruptly in the early 1960s (Figure 1). Infectious disease is now the second leading killer in the world, number three in developed nations and fourth in the United States (The Journal of antibiotics, 70(5), pp.520-526.). For example, current worldwide deaths attributable to antimicrobial resistance have been estimated at ~700,000 per year, rising to 10 million per year by 2050 if present trends continue (Science, 2021, 373(6554), pp.471-471.).
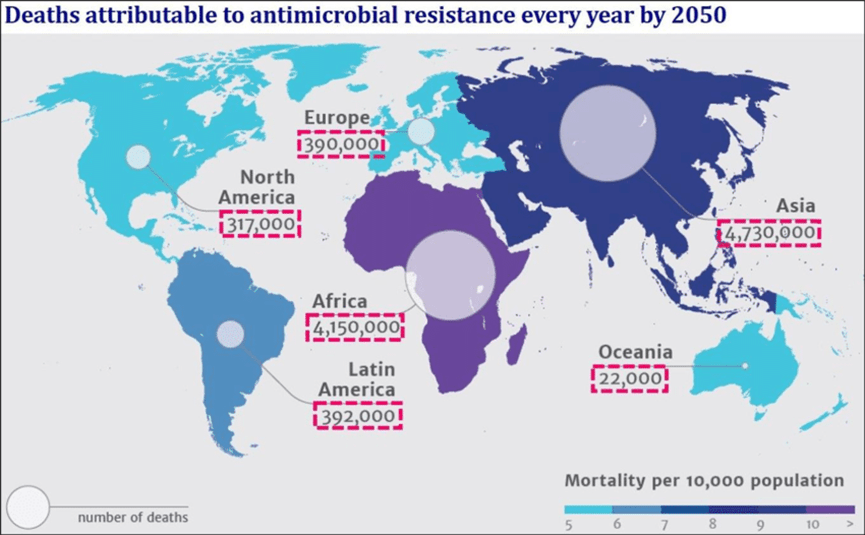
Despite the fact that the spread of antibiotic-resistant bacteria poses a substantial threat to morbidity and mortality worldwide, pharmaceutical research and development has failed to meet the clinical need for new antibiotics. In the past 20 years, only two new antibiotic classes (lipopeptides and oxazolidinones) have been developed and approved by international drug agencies (US Food and Drug Administration and European Medicines Agency)—both of which provide coverage against Gram-positive bacteria. More sadly, the drugs against Gram-negative bacteria have not been developed after the last novel drug, quinolones , discovered in 1962
The World Health Organization (WHO) recently introduced a list of priority pathogens (The Lancet Infectious Diseases, 18(3), pp.318-327.) and noted those of “critical priority”-drug-resistant Enterobacteriaceae (E. coli, Salmonella typhimurium, Klebsiella pneumoniae, Enterobacter), Pseudomonas aeruginosa, and Acinetobacter baumannii, which are responsible for a global health problem. All of the critical priority pathogens are Gram-negative bacteria. The antibiotic resistance crisis threatens the return of the pre-antibiotic era of epidemics and pandemics. It is important to note that antibiotics not only save lives, but enable modern medicine. Without antibiotics, surgery, chemotherapy, or organ transplantation become highly problematic.
Targets of antibiotics targets and resistance mechanisms
Antibiotics are available that effectively inhibit bacterial cell wall synthesis, protein synthesis, and DNA replication (Figure 3a). But bacteria have evolved an array of mechanisms that enable them to resist the inhibitory action of antibiotics. There exists by now an extensive body of knowledge regarding the nature of different mechanistic classes of antibiotic resistance (Figure 3a). Most such mechanisms serve either to reduce antibiotic concentration in the vicinity of the molecular target (through active efflux or chemical inactivation of the antibiotic) or involve a fundamental change in the nature of the target that reduces the antibiotic’s inhibitory effect (for example, as a consequence of mutation or chemical modification). Recent work has revealed that target protection is a key mechanistic player in clinically significant antibiotic resistance that affects diverse classes of antibacterial drugs and is prevalent in bacterial pathogens.
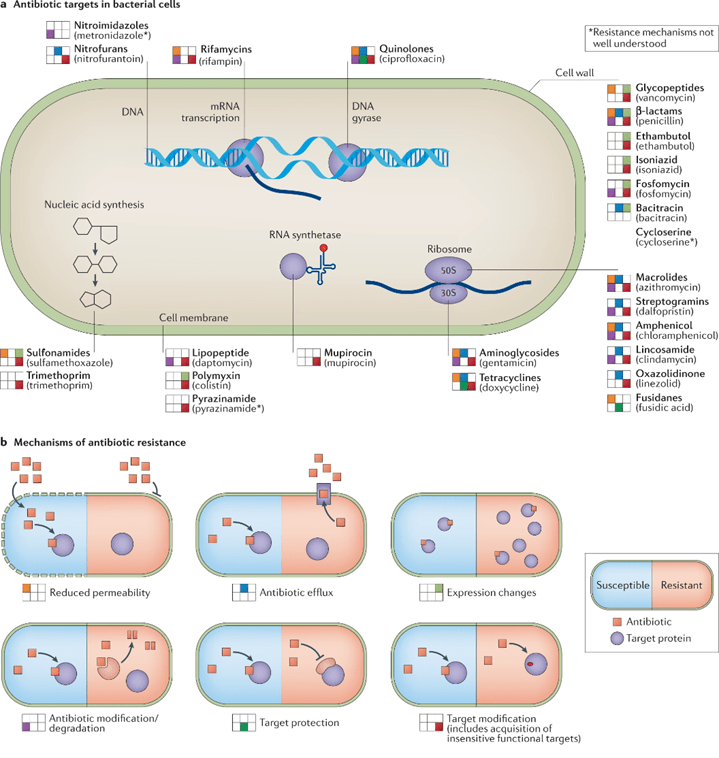
The spread of antibiotic resistance in bacterial pathogens
The emergence of antimicrobial resistant bacteria is not limited to humans or human medicine only, but is influenced by multiple direct and indirect pathways, that form a complex web of interactions that contribute to the selection and spread of resistance in the human/animal microbiota and the environment (Figure 4)
WHO and numerous other groups and researchers agree that the spread of bacterial antimicrobial resistance is an urgent issue requiring a global, coordinated action plan to address(The Lancet. 2022, 399 (10325): 629-655). Understanding how antibiotic resistance evolves and spreads is key to improving antibiotic treatment strategies.
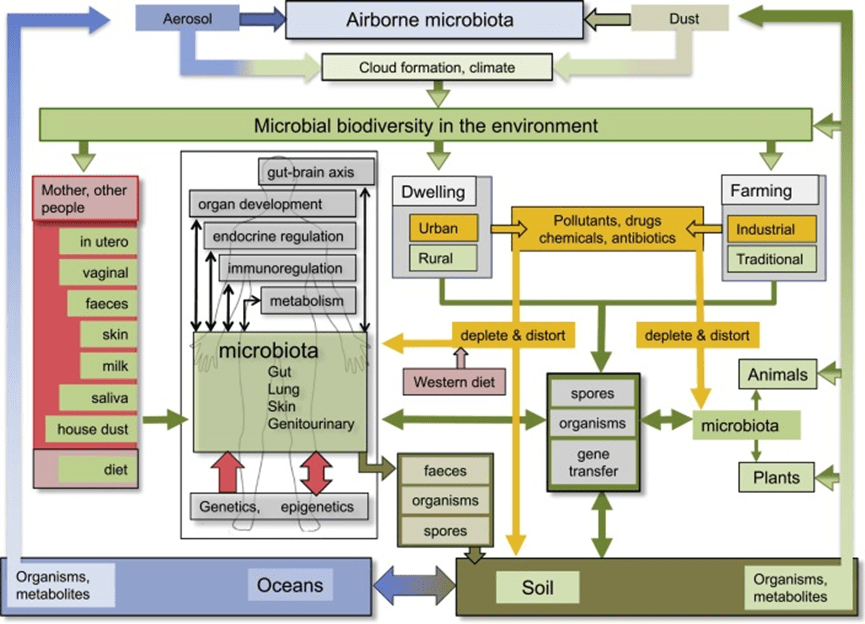
Pathogenic bacteria acquire resistance through two fundamentally different genetic mechanisms: conjugation and transduction (Figure 5). Resistance genes to clinically relevant antibiotics are often carried on plasmids (circular pieces of DNA) that can be transferred between different types of bacteria. Dissemination of drug resistance is mainly due to the ability of bacteria to acquire genes through horizontal transfer mechanisms, primarily by bacterial conjugation. During conjugation, DNA is transferred from a donor to a recipient cell by direct contact. The resulting transconjugant cell will, in turn, disseminate the conjugative DNA, generally as a plasmid that carries all the genes required for its maintenance and transfer. For example, bacteria that are exposed to antibiotics can survive if they have already received a plasmid with an antibiotic resistance gene from another bacterium.
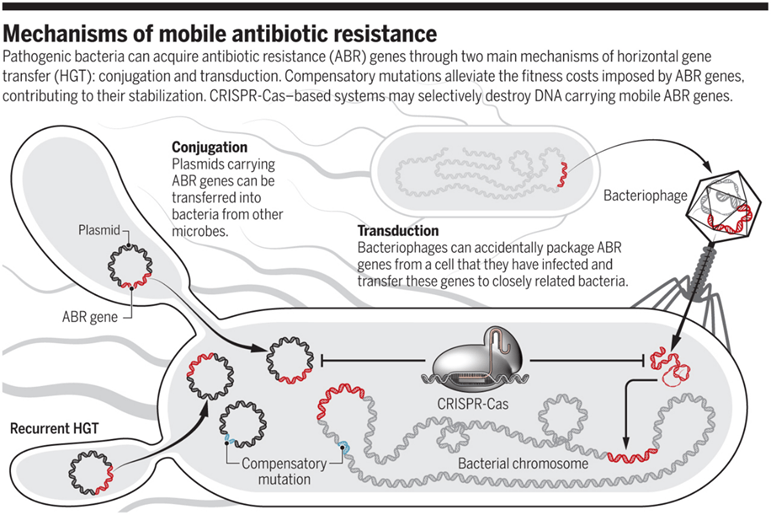
Collaborative action is critical for dealing with antibiotic resistance
COVID-19 has shown that it is possible to create robust public-private partnerships across research, industry, and public health that accelerate research and clinical trials and spur proactive regulation in the context of a global public health threat (Science, 373(6554), pp.471-471.). Collaborative action is equally necessary to battle antimicrobial resistant bacteria.
The scientific community should leverage lessons learned from COVID-19 to unite academia, industry, government, and policy-makers toward preserving the benefits of modern medicine. Continued procrastination will only lead to countless lives lost to antimicrobial resistant bacteria.
References
- Lewis, K., 2020. The science of antibiotic discovery. Cell, 181(1), pp.29-45.
- Martens, E. and Demain, A.L., 2017. The antibiotic resistance crisis, with a focus on the United States. The Journal of antibiotics, 70(5), pp.520-526.
- Kwon JH, Powderly WG. The post-antibiotic era is here. Science. 2021 Jul 30;373(6554):471-.
- Laxminarayan, R., Van Boeckel, T., Frost, I., Kariuki, S., Khan, E.A., Limmathurotsakul, D., Larsson, D.J., Levy-Hara, G., Mendelson, M., Outterson, K. and Peacock, S.J., 2020. The Lancet Infectious Diseases Commission on antimicrobial resistance: 6 years later. The Lancet Infectious Diseases, 20(4), pp.e51-e60.
- Boolchandani, M., D’Souza, A.W. and Dantas, G., 2019. Sequencing-based methods and resources to study antimicrobial resistance. Nature Reviews Genetics, 20(6), pp.356-370.
- Murray, C.J., Ikuta, K.S., Sharara, F., Swetschinski, L., Aguilar, G.R., Gray, A., Han, C., Bisignano, C., Rao, P., Wool, E. and Johnson, S.C., 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet.
- Flandroy, L., Poutahidis, T., Berg, G., Clarke, G., Dao, M.C., Decaestecker, E., Furman, E., Haahtela, T., Massart, S., Plovier, H. and Sanz, Y., 2018. The impact of human activities and lifestyles on the interlinked microbiota and health of humans and of ecosystems. Science of the total environment, 627, pp.1018-1038.
- MacLean, R.C. and San Millan, A., 2019. The evolution of antibiotic resistance. Science, 365(6458), pp.1082-1083.
Production of Semi-synthetic Artemisinin in engineered yeast
Jiang Hao, 17 March 202
1. Artimisinin, a key in the battle against malaria
In 2016, malaria killed an estimated 445,000 people, 90% of them in Africa. That is, one person dies every 71 seconds, caused by preventable and treatable diseases. Despite a 25% drop in deaths since 2010, the World Health Organization recently warned that progress was stalling and called for a redoubled effort to combat the scourge.
Some of the most critical weapons in this battle are artemisinin-based combination therapy (ACT), considered the most effective antimalarial drug. They contain derivatives of artemisinin, a molecule found in the sweet wormwood (Artemisia annua) plant, which grows mainly in China and Vietnam.
But the world supply of artemisinin has been very unstable over the past 15 years. As more farmers grow artemisinin to meet growing demand, the supply of artemisinin has increased and pushed prices down. So, farmers turned to more profitable crops. The supply of artemisinin then went down, and prices soared again. The artemisinin supply roller coaster has hindered efforts to make ACTs available to everyone who needs them.

There are various methods of introducing foreign nucleic acids into a eukaryotic cell: some rely on physical treatment; others rely on chemical materials or biological particles (viruses) that are used as carriers. There are many different methods of gene delivery developed for various types of cells and tissues, from bacterial to mammalian. Generally, the methods can be divided into three categories: physical, chemical, and biological [1].
2. Commericial production of semi-synthetic artemisinin
Alternative sources of artemisinin can solve this problem. Back in 2004, the Bill & Melinda Gates Foundation funded a project to develop a genetically engineered yeast that could make artemisinic acid, a compound just steps away from artemisinin itself. Sanofi eventually commercialized the process in 2013 to produce semi-synthetic artemisinin (SSA), capable of meeting about one-third of global demand.
To reach this achievement, we must fully understand the biosynthetic pathway of artemisinin. It is mainly divided into four parts:
Formation of farnesyl pyrophosphate FPP through both mevalonate pathway (MVA) and non-mevalonate pathway (DXP).
Under the action of amorpha-4,11-diene synthase, FPP is cyclized to form amorphine-4,11-diene, the intermediate of artemisinin.
Under the action of amorpha-4,11-diene oxidase, amorpha-4,11-diene is further oxidized to form artemisinol and artemisinaldehyde, and then artemisinic acid are synthesized.
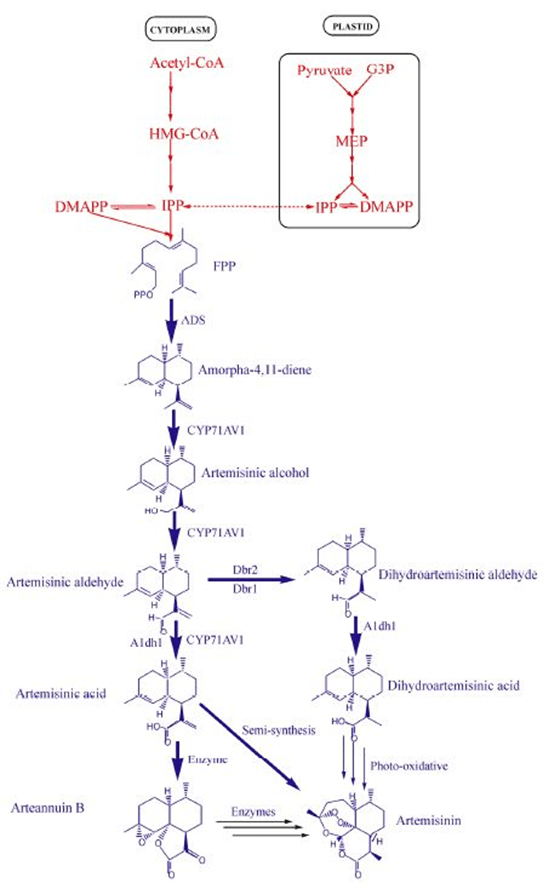
In short, beer yeast (Saccharomyces cerevisiae) was designed to overexpress both the enzyme of the mevalonate pathway and A. annua amorphadiene synthase, resulting in over 40 g/L of amorphadiene (a hydrocarbon precursor of artemisinic acid) in fed-batch fermenters fed with ethanol. Cytochrome P450 enzymes (CYP71AV1) and their homologous reductase (AaCPR) are responsible for the oxidation of amorphadiene, but their expression in yeast results in low conversion of amorphadiene to artemisinic acid. High-level production of artemisinic acid (25 g/L) by yeast fermentation on a 2 L scale was achieved by reducing AACRP expression to alter the stoichiometry of CYP71AV1:AaCPR interactions, as well as co-expression of other enzymes (Cytochrome b5 and two dehydrogenases) are involved in three oxidation reactions that convert amorphadiene to artemisinic acid. Commercial production of semi-synthetic artemisinin began in 2013 with the development of an industrial process for the chemical conversion of artemisinin to artemisinin.
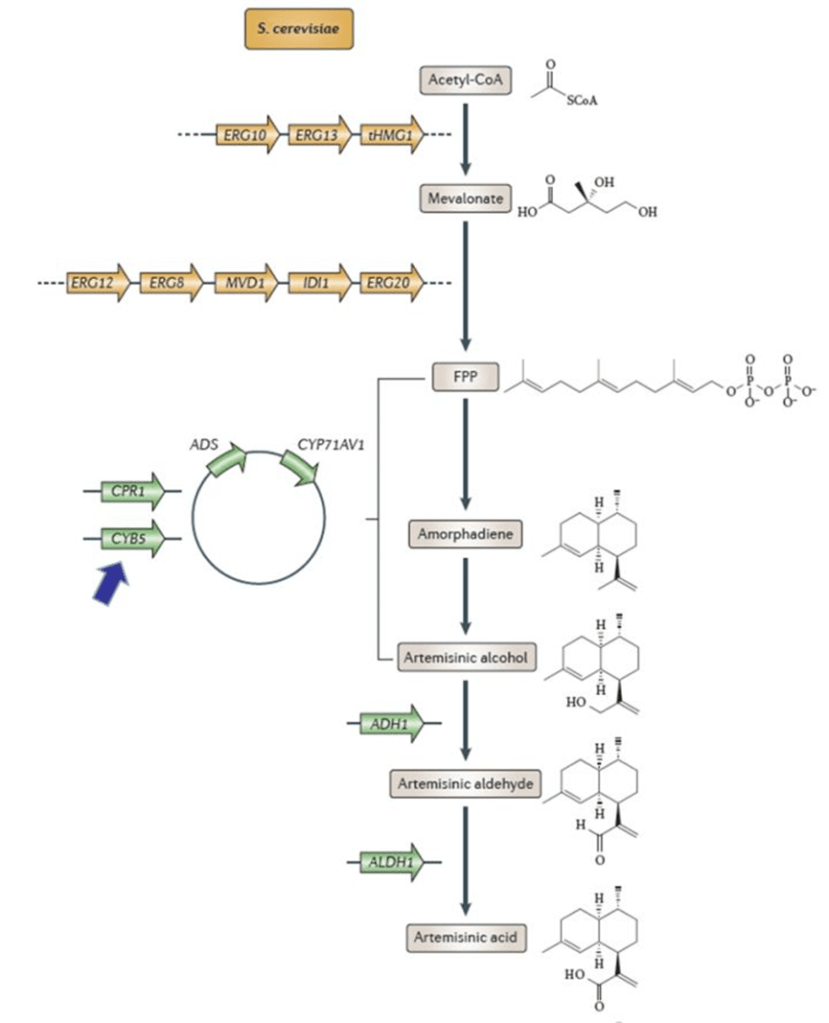
The SSA accounted for about 8% of the global supply of artemisinin in 2013, but fell to zero in 2015. It’s mainly because of its relatively high costs and other ACT manufacturers were reluctant to buy SSA from its rival Sanofi. The following year, sanofi sold its Garesio plant to Huvepharma. There is now a search for new ideas that could “provide a truly sustainable supply of low-cost semi-synthetic artemisinin.”
3. Recent development in increasing the activity of Cryp71Av1
At current production levels, the concentration of amorphadiene in the fermenter is close to 200 mM, well beyond the solubility limit. While CYP71AV1 is able to achieve such high conversions at extremely high concentrations of amorphadiene, methods are still needed to design improved P450 conversions and yield higher, economically relevant artemisinic acid titers.
Direct engineering of catalytic systems can be accomplished in several ways. Based on previous success in titrating AaCPR and cytochrome b5 expression levels, we know that regulation of enzymes indirectly involved in catalysis can have dramatic effects. In addition to designing the active site of P450, engineering how P450 interacts with the yeast endoplasmic reticulum has been fruitful for efforts such as heterologous hydrocodone production. Manipulation of the yeast genome may also be a means of increasing heterologous P450 activity, for example, it was recently shown that a mutation that causes yeast endoplasmic reticulum expansion (Δpah1) results in increased heterologous production of triterpenoid saponins.
References
- Peplow, M. “Looking for cheaper routes to malaria medicines.” Chem Eng News 96.11 (2018): 29-31.
- Ro, Dae-Kyun, et al. “Production of the antimalarial drug precursor artemisinic acid in engineered yeast.” Nature 440.7086 (2006): 940-943.
- Kung, Stephanie H., et al. “Approaches and recent developments for the commercial production of semi-synthetic artemisinin.” Frontiers in plant science 9 (2018): 87.
- Arendt, Philipp, et al. “An endoplasmic reticulum-engineered yeast platform for overproduction of triterpenoids.” Metabolic engineering 40 (2017): 165-175.
- Tu, Youyou. “Artemisinin—a gift from traditional Chinese medicine to the world (Nobel lecture).” Angewandte Chemie International Edition 55.35 (2016): 10210-10226.
- Hale, Victoria, et al. “Microbially derived artemisinin: a biotechnology solution to the global problem of access to affordable antimalarial drugs.” The American journal of tropical medicine and hygiene 77.6_Suppl (2007): 198-202.
- Paddon, Chris J., and Jay D. Keasling. “Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development.” Nature Reviews Microbiology 12.5 (2014): 355-367.
Chemical-based Transfection and Its Applications in Vaccines
Li Biquan, 10 March 2022
1. Introduction to Transfection
Transfection, which is the process of introducing foreign nucleic acids into host cells, is one of the most powerful molecular biology tools currently in use. In animal cells, transfection is the preferred term. Additionally, we should pay attention to the differences of the concept between transfection, transformation, and transduction.
Generally, we can divide transfection into categories, stable and transient transfection, which differ in their long-term effects on a cell. A stably-transfected cell will continuously express transfected DNA and pass it on to daughter cells, while a transiently-transfected cell will express transfected DNA for a short amount of time and not pass it on to daughter cells. From Figure 1, we could compare the differences between stable and transient transfection, and we can note that in the process of stable transfection, the DNA would be integrated into the gene of the host cell, while transient transfection doesn’t experience this step [1].
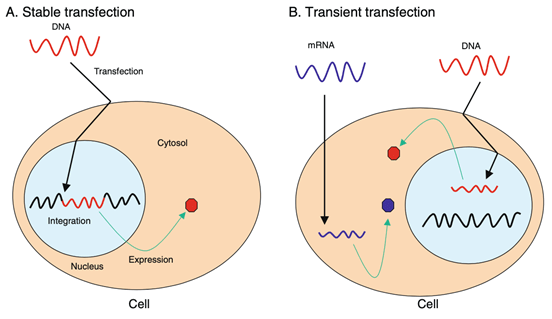
There are various methods of introducing foreign nucleic acids into a eukaryotic cell: some rely on physical treatment; others rely on chemical materials or biological particles (viruses) that are used as carriers. There are many different methods of gene delivery developed for various types of cells and tissues, from bacterial to mammalian. Generally, the methods can be divided into three categories: physical, chemical, and biological [1].
2. Chemical-based Transfection
In this abstract, we are mainly to introduce chemical-based transfection. As shown in Figure 2, Chemical-based transfection can be divided into several kinds: calcium phosphate, cationic polymers, liposomes (lipofection) and, nanoparticles etc [1].
One of the cheapest methods uses calcium phosphate, originally discovered by F. L. Graham and A. J. van der Eb in 1973 [2]. This process has been a preferred method of identifying many oncogenes and proved that the complex access to the cell through endocytosis. However, the transfection efficiency strongly depends on the cell constitution, the pH, and the quality and the amount of the used nucleotides, and it is harmful and therefore not suitable for most sensitive primary cell lines [1].
Another method is the use of cationic polymers such as DEAE-dextran or polyethylenimine (PEI). The negatively charged DNA binds to the polycation and the complex is taken up by the cell via endocytosis. This method can be transiently transfected into eukaryotic cells in an inexpensive and simple manner and is able to be easily reproduced, but the efficiency in a number of cell types is low (including primary cells) and it is cytotoxic and therefore not suitable for sensitive cells [3].
Lipofection (or liposome transfection) is a technique used to inject genetic material into a cell by means of liposomes, which are vesicles that can easily merge with the cell membrane since they are both made of a phospholipid bilayer [4]. Lipofection generally uses a positively charged (cationic) lipid (cationic liposomes or mixtures) to form an aggregate with the negatively charged (anionic) genetic material [5]. This transfection technology performs the same tasks as other biochemical procedures utilizing polymers, DEAE-dextran, PEI and, calcium phosphate [6]. This method is easy to apply and yields highly reproducible results of transient and stable transfections, but the transfection efficiency strongly depends on the cell type. Another disadvantage is that he viability after the transfection process might be decreased due to the high cellular sensitivity, especially for primary and non-dividing cells. It is worth noting that lipofection is based on endosomal molecule uptake, and also can easily merge with the cell membrane since they are both made of a phospholipid bilayer [5].

The last method we’d like to introduce and use in vaccines is nanoparticle-based transfection. Some cells are relatively easy to transfect, whereas others, such as stem cells, suspension cells, or primary cells show poor transfection results. This is where nanoparticles, each behaving as a single unit, come into the play, owing to their ultra-miniaturized size (ranging from 1 to 100 nm) [1, 7]. This method is used in many examples, such as AuNPs-based CRISPR delivery [8] and LNPs-based mRNA (vaccine) delivery [9, 10], which is shown in Figure 3.
LNP-based delivery is one of the most and main ways in mRNA vaccines delivery, other major delivery methods for mRNA vaccines Commonly used delivery methods and carrier molecules for mRNA vaccines are shown in Figure 4. [11]
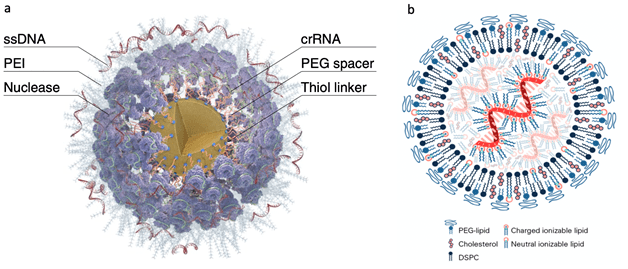
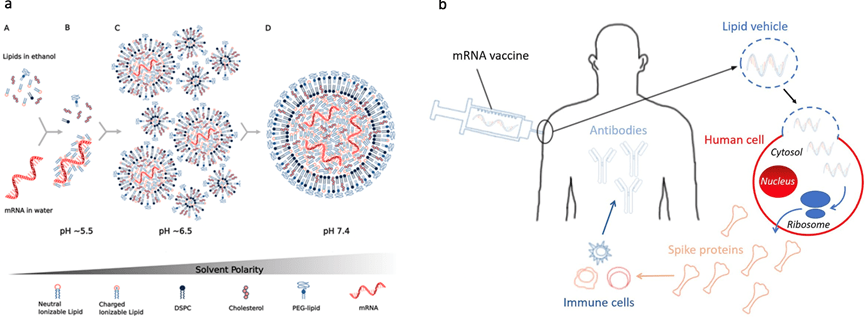
5. Assembly of mRNA Lipid Nanoparticles (Vaccines)
The current methods of mRNA lipid nanoparticle assembly is achieved by rapid mixing in a microfluidic or T-junction mixer of four lipids (ionizable lipid, DSPC, cholesterol, PEG–lipid) in ethanol with mRNA in an aqueous buffer near pH is 4. When the ionizable lipid meets the aqueous phase, it becomes protonated at a pH ~5.5, which is intermediate between the pKa of the buffer and that of the ionizable lipid. The ionizable lipid then electrostatically binds the anionic phosphate backbone of the mRNA while it experiences hydrophobicity in the aqueous phase, driving vesicle formation and mRNA encapsulation. After initial vesicle formation, the pH is raised by dilution, dialysis or filtration, which results in the neutralization of the ionizable lipid, rendering it more hydrophobic and thereby driving vesicles to fuse and causing the further sequestration of the ionizable lipid with mRNA into the interior of the solid lipid nanoparticles. The PEG–lipid content stops the fusion process by providing the LNP with a hydrophilic exterior, determining its thermodynamically stable size, and the bilayer forming DSPC is present just underneath this PEG–lipid layer [10]. (shown in Figure 5 (a))
6. COVID-19 mRNA Vaccination Mechanism
Initially, the mRNA vaccine is injected by intramuscular route, typically into the deltoid muscle. Subsequently, the lipid coat vehicle (lipid nanoparticles) around the mRNA allows for the vaccine to enter the cytosol of the cell, typically Dendritic cells (DCs), antigen-presenting cells. And then, the ribosomes translate the mRNA into spike proteins, and the injected mRNA subsequently degrades. The spike proteins are released from the cell and initiate an adaptive immune response. Eventually, through various activation pathways, immune cells mount a cell-mediated and antibody-mediated immunity against the spike protein of the SARS-CoV-2 virus [12]. (shown in Figure 5 (b))
References
[1] Kim TK, Eberwine JH.Mammalian cell transfection: the present and the future. Analytical and Bioanalytical Chemistry. Anal Bioanal Chem, 2010, 397: 3173-3178.
[2] Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973, 52 (2): 456-67.
[3] Christina Ehrhardt, Mirco Schmolke, Andreas Matzke, Alexander Knoblauch, Carola Will, Viktor Wixler, and Stephan Ludwig. Polyethylenimine, a cost-effective transfection reagent. Signal Transduction, 2006, 6: 179-184.
[4] Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proceedings of the National Academy of Sciences of the United States of America. 1987, 84 (21): 7413-7.
[5] Felgner JH, Kumar R, Sridhar CN, Wheeler CJ, Tsai YJ, Border R, Ramsey P, Martin M, Felgner PL. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. The Journal of Biological Chemistry. 1994, 269 (4): 2550-61.
[6] P L Felgner, T R Gadek, M Holm, R Roman, H W Chan, M Wenz, J P Northrop, G M Ringold, M Danielsen. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Nati. Acad. Sci., 1987, 84: 7413-7417.
[7] Rein Verbeke, Ine Lentacker, Stefaan C. De Smedt, Heleen Dewitte. Three decades of messenger RNA vaccine development. Nano Today, 2019, 28: 100766.
[8] Reza Shahbazi, Gabriella Sghia-Hughes, Jack L. Reid, Sara Kubek, Kevin G. Haworth, Olivier Humbert, Hans-Peter Kiem and Jennifer E. Adair. Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nature Materials, 2019, 18, 1124-32.
[9] Na-Na Zhang, Xiao-Feng Li, Yong-Qiang Deng, You-Chun Wang, Bo Ying, Cheng-Feng Qin. A Thermostable mRNA Vaccine against COVID-19. Cell, 2020, 182, 1271-83.
[10] Michael D. Buschmann, Mohamad Gabriel Alameh, Manuel J. Carrasco, Suman Alishetty, Mikell Paige, and Drew Weissman. Vaccines, 2021, 9 (65): 1-30.
[11] Yang Wang, Ziqi Zhang, Jingwen Luo, Xuejiao Han, Yuquan Wei and Xiawei Wei. mRNA vaccine: a potential therapeutic strategy. Mol Cancer, 2021, 20 (33): 1-23.
[12] Pratibha Anand and Vincent P. Stahel. The safety of Covid-19 mRNA vaccines: a review. Anand and Stahel Patient Safety in Surgery, 2021, 15 (20):1-9.
Proximity labeling: a new tool to explore molecular interactions
Zhang Yu, 17 Feb. 2022
Many biological processes are regulated through the molecular interactions of proteins and nucleic acids. In the past several years, affinity purification and yeast two-hybrid have been widely applied to discover potential molecular interactions. (1,2) Antibody-based affinity purification, in combination with mass spectrometry–based proteomics, allows the enrichment and identification of stable interaction partners of specific proteins of interest. The main limitation of affinity purification, however, is that weak or transient interactions are often lost during cell lysis and the subsequent washing steps. Moreover, affinity purification is challenging to apply to insoluble targets or protein baits.
Yeast two-hybrid and other protein complementation assays represent another approach for mapping protein–protein, protein–RNA and protein-DNA interactions in living cells. (1) These approaches are often high throughput, enabling the screening of thousands to millions of potential molecular interactions. (3) However, many proteins complementation assays have cell type and organelle type restrictions.

Then, with the development of the molecular biology, a brand-new technology was developed—the proximity labelling. Proximity labeling (PL) is a technology used to find the interaction partners of ‘baits’ proteins by fusing ‘baits’ proteins with enzymes who can modified proximity molecules. Tagged molecules that interact with baits can then be enriched and identified by mass spectrometry or nucleic acid sequencing. (4) The predecessor of the proximity labelling is DamID. It is a method developed in 2000 by Steven Henikoff for identifying parts of the genome proximal to a chromatin protein of interest. (5) DamID relies on a DNA methyltransferase fusion to the chromatin protein to nonnaturally methylate DNA, which can then be subsequently sequenced to reveal genome methylation sites near the protein. Researchers were guided by the fusion protein strategy of DamID to create a method for site-specific labeling of protein targets, culminating in the creation of the biotin protein labelling-based BioID in 2012. (6) Alice Ting and the Ting lab at Stanford University have engineered several proteins that demonstrate improvements in biotin-based proximity labeling efficacy and speed.
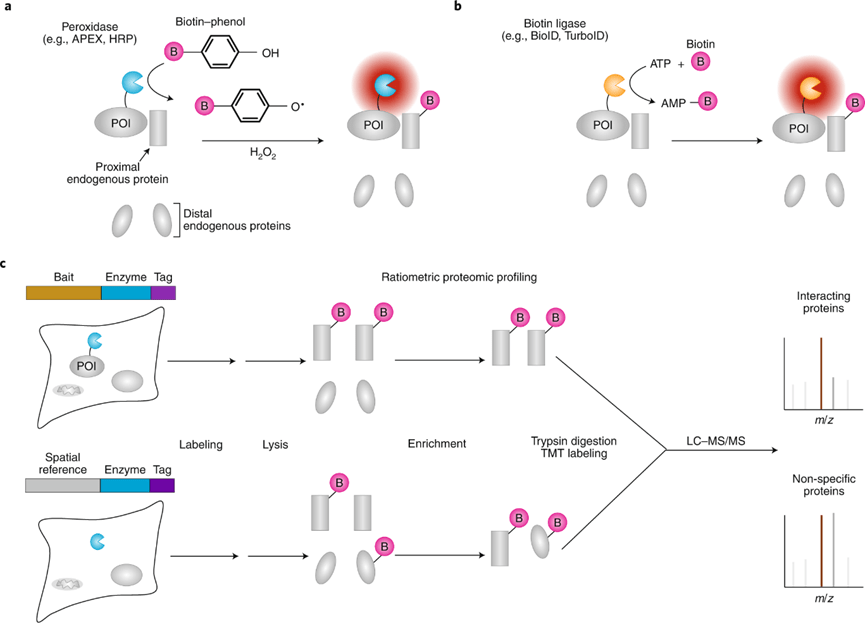
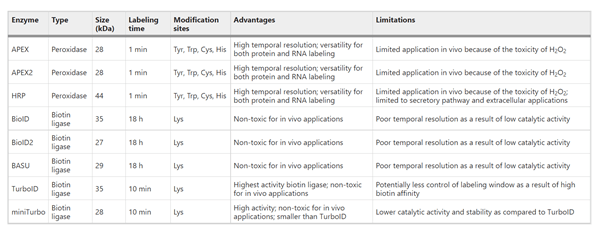
For now, several enzymes were selected as tagging enzymes to fuse with the ‘baits’ proteins. (Table 1) Each of them has its own advantages and disadvantages according to the application scenario. Just take APEX for an example, the requirement of the toxic H2O2 and the short labelling time make it a good option in the dead cells experiments. BASU provides a possible tagging scheme in living cells with a disadvantage of long labelling time. None of them can be applied to all situations. Besides, avoiding background from endogenously biotinylated proteins to improve accuracy for PL in vivo and developing more PL enzymes which will boost sensitivity and analysis of transcriptomes and genomes in distinct cell populations is still a long but meaningful work for researchers.
In conclusion, proximity labelling is a burgeoning field with lots of chances and challenges. Continuing development of increasingly sophisticated PL technology may vastly expand the range of PL-based discoveries and address more challenging questions, such as determining the affinity, stoichiometry and contact sites of molecular interactions.
References
1. Brückner, A., Polge, C., Lentze, N., Auerbach, D. & Schlattner, U. Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 10, 2763–2788 (2009).
2. Dunham, W. H., Mullin, M. & Gingras, A. C. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics 12, 1576–1590 (2012).
3. Trigg, S. A. et al. CrY2H-seq: a massively multiplexed assay for deep-coverage interactome mapping. Nat. Methods 14, 819–825 (2017).
4. Qin, W., Cho, K. F., Cavanagh, P. E., Ting, A. Y., Nat. Methods 18, 133-143 (2021).
5. Steensel, B., Henikoff, S. Identification of in vivo DNA targets of chromatin proteins using tethered Dam methyltransferase. Nat Biotechnol 18, 424–428 (2000).
6. Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 196, 801-810 (2012).
Potential of AlphaFold in Determining Protein Structure
Han Yongxu, 10 Feb. 2022
Centra dogma uncovers the production of protein which was translated from mRNA caring the genetic information of DNA. Protein-related research is the most fertile areas in chemical biology and medical biology. Function of protein is determined by its conformation which is complicated and variable, thus deciphering how protein worked entails the illustration of the structure.
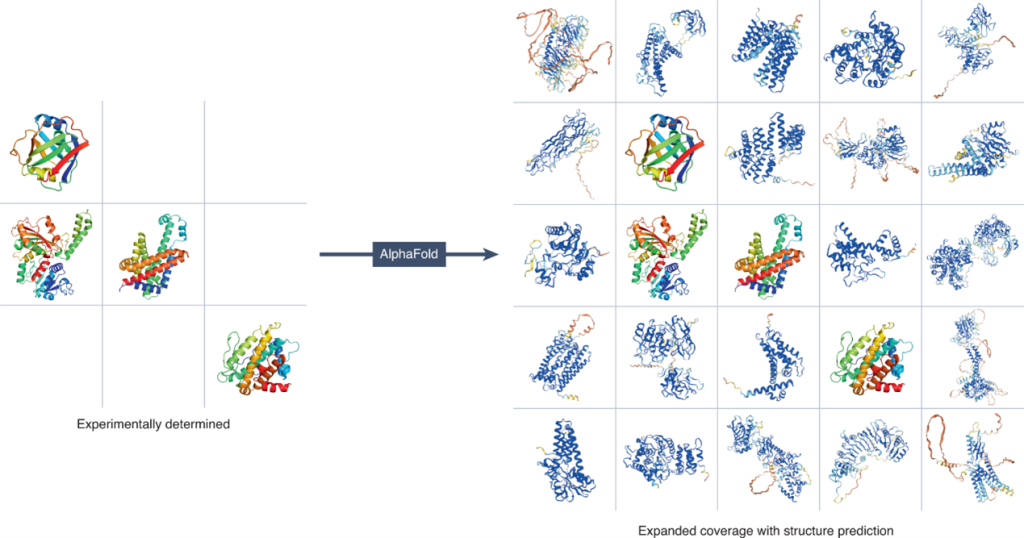
The determination of protein structures can be dated from 60 years ago. John Kendrew, as a biologist from England, was awarded the Nobel Prize in chemistry in 1962 due to the contribution on the determination of myoglobin which containing heme with x-ray crystallography. Several scientist were also granted the Nobel Prize for the discovery on the conformation of proteins like virus particles, rhodopsin protein, aquaporins and ion channel protein and so forth in the following years. X-ray diffraction crystallography, NMR and cryo-EM were major methods to explore the protein structures. Searching from the Protein Data Bank (PDB), we can found that there are about 180,000 released entries for protein structures, among of them, more than 80% were determined from X-ray crystallography, 13,448 were solved by NMR and no more than 10, 000 were from cryo-EM (1). X-ray diffraction crystallography was one of the first experimental methods used for structural resolution, however, X-ray diffraction cannot be applied to larger proteins (2). NMR was generally believed to be able to describe the real structure of proteins in solutions rather than crystal structure. Moreover, NMR analysis can obtain the structural position of hydrogen atom. However, due to the unstable structures of proteins in solution sometimes, it is difficult to capture stable signals, thus computer modeling and other methods are supplemented for the structure determination (3). Cryo-EM method in resolving bound ligands and delineating structured water molecules makes it possible to obtain detailed information relevant to molecular mechanisms (4).
AlphaFold known from Critical Assessment of Structure Prediction (CASP) conference is a neural-network-based approach to predicting protein structures with high accuracy. Recently, the source code and almost 350,000 protein models within AlphaFold 2 from various species, including human and bacterial have been made public (5).
The network of AlphaFold can directly predict 3D coordinates of all heavy atoms for a given protein using the primary amino acid sequence and aligned sequences of homologues as inputs, then apply multiple sequence alignments (MSAs) and pairwise features to illustrate a new architecture. There are two index including the predicted local-distance difference test (pLDDT) and template modelling score (TM-score) to judge the prediction efficiency. pLDDT can reliably predict the Cα local-distance difference test (lDDT-Cα) accuracy of the corresponding prediction, and the latter one presents the global superposition metric template modelling score (6).
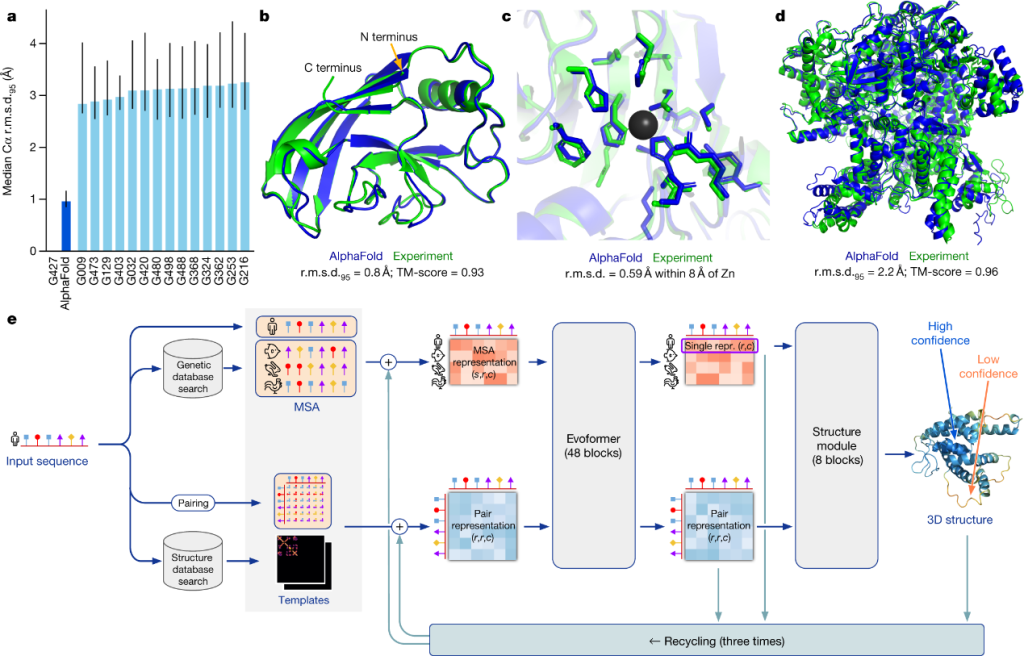
Apart from the predication of unknown structures of proteins, AlphaFold can also predict certain classes of protein–protein interfaces (7). Researchers have established 3D models for 106 previously unidentified complexes and 806 that have not been structurally characterized from yeast with the combination of RoseTTAFold and AlphaFold (8). In addition, one report on drug discovery by Alphafold have been published. The authors shown that they can screen a target molecule to CDK20 for the treatment of HCC within 30 days combining the AI engines PandaOmics and Chemistry42 (9).
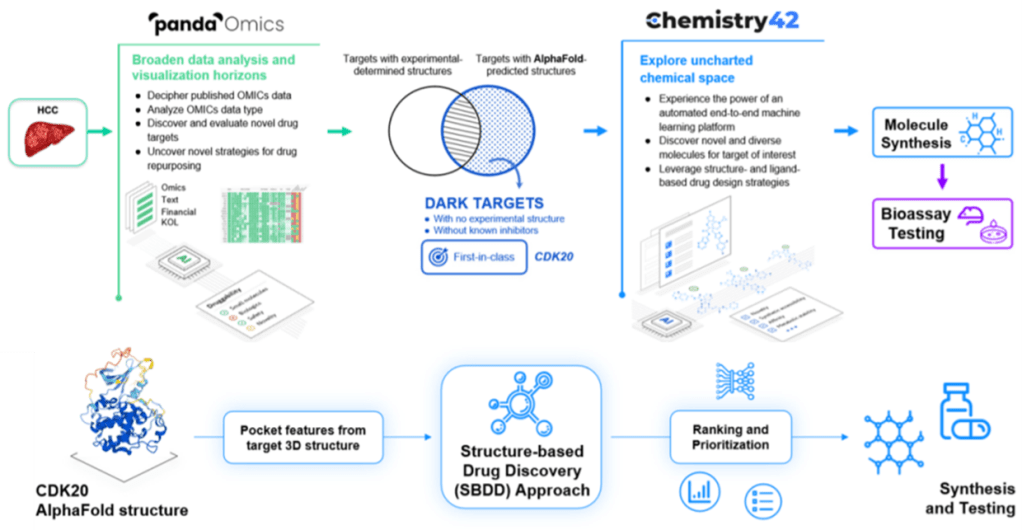
In conclusion, AlphaFold 2 open the door to the application for protein conformational predication with integrating novel neural network architectures and training processes according to the evolutionary, physical and geometric constraints of protein structures (6,10). It is undoubtedly that AlphaFold 2 makes contribution to the medicine discovery and clarification of protein interaction which is related to the cell process at molecular pathway, and thus giving perspective of biological function. However, some scientists think that the protein-folding problem by AlphaFold 2 has not been solved yet. In addition, AlphaFold 2 cannot predicate the complicated protein structures in the dynamic process which is universal in a real cell. Proteins in allosteric states where it is bonded to partners like ligand, cofactor, DNA or other macromolecules can varied dramatically, thus it is difficult to predicate the accurate protein structures at specific moment. Although innovative work are entailed to address the mentioned above limitations, the introduction of AlphaFold 2 herald a new era in the development of structural biology and medical biology.
References
- Subramaniam, Sriram, and Gerard J. Kleywegt. A paradigm shift in structural biology. Nature methods 19.1 (2022): 20-23.
- Schotte, Friedrich, et al. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science 300.5627 (2003): 1944-1947.
- Sakakibara, Daisuke, et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 458.7234 (2009): 102-105.
- Yip, Ka Man, et al. Atomic-resolution protein structure determination by cryo-EM. Nature 587.7832 (2020): 157-161.
- Tunyasuvunakool, Kathryn, et al. Highly accurate protein structure prediction for the human proteome. Nature 596.7873 (2021): 590-596.
- Jumper, John, et al. Highly accurate protein structure prediction with AlphaFold. Nature 596.7873 (2021): 583-589.
- Baek, Minkyung, et al. “Accurate prediction of protein structures and interactions using a three-track neural network.” Science 373.6557 (2021): 871-876.
- Humphreys, Ian R., et al. Computed structures of core eukaryotic protein complexes. Science 374.6573 (2021): eabm4805.
- Ren, Feng, et al. AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel Cyclin-dependent Kinase 20 (CDK20) Small Molecule Inhibitor. arXiv preprint arXiv:2201.09647 (2022).
- Senior, Andrew W., et al. Improved protein structure prediction using potentials from deep learning. Nature 577.7792 (2020): 706-710.
Protein Liquid-Liquid Phase separation
Liu Min, 20 Jan. 2022
Since 2009, when Hyman’s group discovered the liquid property of P granules from C. elegans, most of the research hotspots focused on the protein condensate. Actually, since 2000, many scientists have discovered various membrane-less structures. Most of these structures are 100-1000 nm diameters and contain a large amount of protein and RNA, which are involved in the normal physiological activities of cells. For example, stress granules are structures associated with translation regulation, while dvl2 puncta are associated with cell signaling.
Liquid-liquid separation is a very common phenomenon in the field of physical chemistry. Many substances are single phase in the monomer stage, and when polymerized they form two phases. The protein phase separation is more complicated than polymer, and the two most important driving forces are the disorder of the protein sequence and the multivalent effect. For example, simply increasing the concentration of a protein component can see phase separation, increasing the valency of a component can also promote phase separation, or reducing the solubility of a component can also help phase separation.
In 2012, Rosen’s group from Southwestern Medical Center reconstituted the phase-separated system for the first time in vitro. They successfully realized the observation of phase-separated droplets by increasing the valency of the two interacting proteins. Afterwards, Professor Clifford P. Brangwynne from Princeton did a lot of interesting work on the phase separation system from discovery, establishment to application, which are related to the transcription and translation of genes.
Phase-separated systems have now expanded into many applications. For example, in enzymatic reactions, it can be used as a concentrated enzyme and substrate to facilitate the rapid progress of the reaction. In addition, in signal transduction, it can function as a sequestration to achieve organelle-like functions. There are also many people who have made some organizational hubs to achieve some separated functions through a phase-separated system.
In conclusion, phase separation is a complex system integrating biology, physics and chemistry, and its functions and applications are still being explored. In future research, we expect more valuable discoveries and phenomena.
References
- Brangwynne, Clifford P., et al. “Germline P granules are liquid droplets that localize by controlled dissolution/condensation.” Science 324.5935 (2009): 1729-1732.
- Li, Pilong, et al. “Phase transitions in the assembly of multivalent signalling proteins.” Nature 483.7389 (2012): 336-340.
- Feric, Marina, et al. “Coexisting liquid phases underlie nucleolar subcompartments.” Cell 165.7 (2016): 1686-1697.
- Kedersha, Nancy, and Paul Anderson. “Mammalian stress granules and processing bodies.” Methods in enzymology 431 (2007): 61-81.
- Schwarz-Romond, Thomas, Ciara Metcalfe, and Mariann Bienz. “Dynamic recruitment of axin by Dishevelled protein assemblies.” Journal of cell science 120.14 (2007): 2402-2412.
- Shin, Yongdae, and Clifford P. Brangwynne. “Liquid phase condensation in cell physiology and disease.” Science 357.6357 (2017).
The virulence mechanisms of EPEC
Qiu Jiaming, 13 Jan. 2022
Enteropathogenic Escherichia coli (EPEC) is an attaching and effacing (A/E) pathogen, which can cause significant paediatric morbidity and mortality all over the world, especially in developing countries. The infection of EPEC involves four stages: attachment, sending in effector proteins, actin reorganization, and pedestal formation and includes various elements which play a vital role in EPEC infection.
T3SS
Firstly, EPEC uses locus of enterocyte effacement (LEE) pathogenicity island, which encodes a type III secretion system (T3SS) and various cognate effectors (Tir, Map, EspF, EspG, EspH, and EspI) to infect host cells. Through the integration of coordinated intracellular and extracellular cues, the T3SS is assembled within the bacterial cell wall, as well as the plasma membrane of the host cell. T3SS construction includes four stages: (i) assembly of the basal body and export apparatus, (ii) assembly of the inner rod and needle, (iii) assembly of the filament and translocon, and (iv) secretion of effectors. And along with the formation of injectisome, EPECs can complete the infection.
Intimin and Tir
Secondly, the especially feature of A/E bacterial pathogens is the formation of pedestal beneath tightly adherent bacteria and localized disruption of the brush border of the intestine. And in this process, the intimin which belongs to bacterial outer membrane adhesin, is required for the production of A/E lesions and diarrhea. As we known, EPEC use T3SS to translocate their own intimin receptor, Tir, to the membranes of mammalian cells. The translocated Tir triggers additional host signaling events and actin nucleation. Actin nucleation is critical for lesion formation. Current studies have demonstrated that when the IBD domain of Tir binds to the D3 domain of the EPEC intimin protein on the mammalian cell surface, the bacteria are primed to remain on the mammalian cell surface.
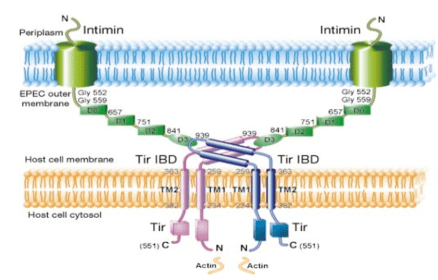
N-WASP
N-WASP is best known as an Arp2/3 complex activator in processes like actin nucleation and has been reported to regulate actin nucleation and polymerization for multiple cell activities. It was found that at the carboxyl terminus of N-WASP, there is a conserved VCA region, which is composed of the verprolin homology region (V), the cofilin homology region (C) and an acidic region (A). The acidic region and the cofilin homology region bind to the actin-associated complex Arp2/3, and upon binding, promote the conversion of F-actin to G-actin, which will eventually cause the polymerization of actin leading to the formation of pedestal.The amino-terminal region of N-WASP has an EVH1 (WH1) structural domain immediately followed by a guanosine triphosphatase (GTPase) binding domain. The GTP hydrolase binding domain binds to the GTPase Cdc42 and can activate the function of N-WASP. Between the amino- and carboxy-terminal structural domains of N-WASP is a proline-rich sequence, and this structural domain can bind to the Src homology (SH)3 structural domains of signaling proteins such as Grb2 and Nck, as well as cytoplasmic tyrosine kinases.

In conclusion, EPEC infection is an extremely complex physiological and biochemical process, citing various proteins and physiological processes such as T3SS, Intirmin, Tir, Nck, N-WASP, Arp2/3, and actin, all of which play an important and irreplaceable role in the EPEC infection process. It is also only when we fully understand the functions and mechanisms of these proteins and interactions that we can more effectively research antibacterial drugs and contribute to human health.
References
- Todd, E. C. Epidemiology of foodborne diseases: a worldwide review. World Health Stat Q. 1997;50(1-2)
- Yu Luo et al. Crystal structure of enteropathogenic Escherichia coli intimin–receptor complex. Nature. 2000 Jun 29;405(6790)
- Sabrina L Slater et al. The Type III Secretion System of Pathogenic Escherichia coli. Curr Top Microbiol Immunol. 2018;416(51-72).
- Neal M Alto et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell. 2006 Jan 13;124(1)
- Teemu Kallonen , Christine J Boinett. EPEC: a cocktail of virulence. Nat Rev Microbiol. 2016 Apr;14(4):196.
- J Fawcett , T Pawson. AN-WASP Regulation–the Sting in the Tail. Science. 2000 Oct 27;290(5492):725-6
Bioorthogonal reactions
Yuan Dingdong, 22 Dec. 2021
Bioorthogonal chemistry means any chemical reaction that may occur inside living systems without interfering with native biochemical processes. This concept was coined by Carolyn R. Bertozzi in 2003[1]. The applications of bioorthogonal chemistry are diverse and include genetic code expansion and metabolic engineering, drug target identification, antibody–drug conjugation and drug delivery. Key reactions include native chemical ligation and the Staudinger ligation, copper-catalysed azide–alkyne cycloaddition, strain-promoted [3 + 2] reactions, tetrazine ligation, metal-catalysed coupling reactions, oxime and hydrazone ligations as well as photoinducible bioorthogonal reactions (Fig. 2) [2].
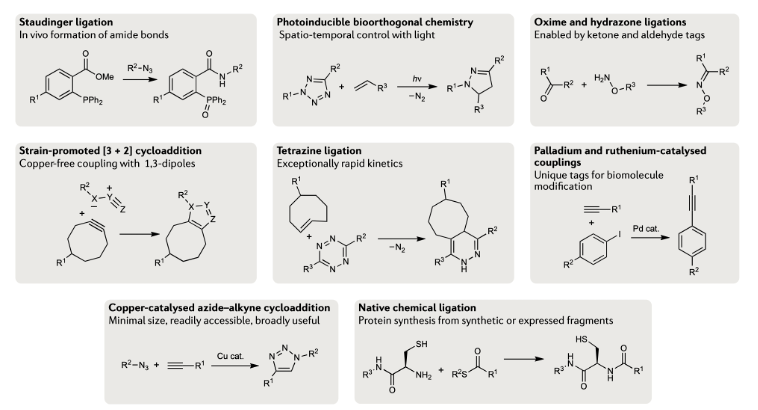
Amongst all bioorthogonal reactions developed to date, the [4+2] cycloaddition of 1,2,4,5-tetrazines (s-tetrazines, Tz) and various dienophiles, referred as inverse electron demand Diels–Alder (IEDDA) reaction, is the one that satisfies most of the bioorthogonal criteria (e.g., fast, selective, biocompatible and catalyst-free) necessary for use in applications from protein labelling to cancer imaging or materials science. Diels–Alder [4+2]-cycloaddition describes the reaction between a diene (e.g., 1,2,4,5-tetrazines) and a dienophile (alkene or alkyne) to form a six-membered ring in a π4s + π2s fashion, via suprafacial/suprafacial interaction of 4π-electrons of the diene with the 2π-electrons of the dienophile (Fig. 3) [3].
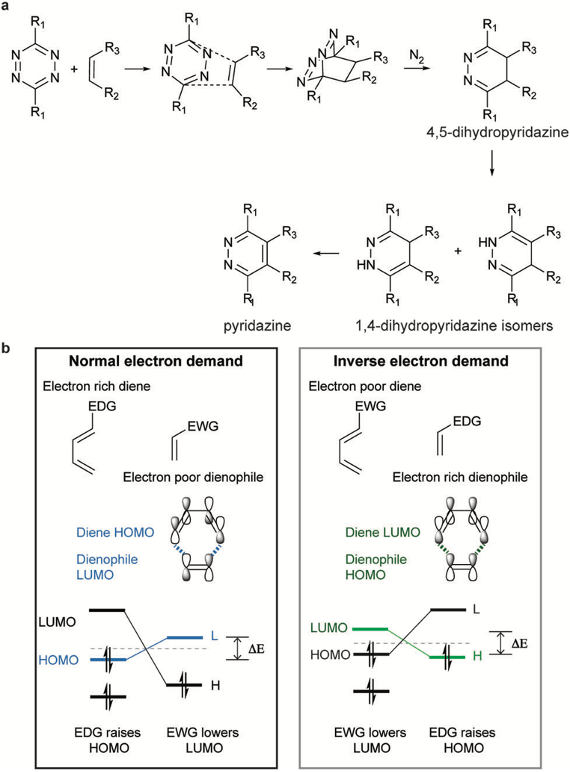
Tetrazine can react with rich and varied strained or unstrained dienophiles, such as trans-cyclooctene (TCO), cyclopropane, norbornene and styrene which can be tuned to reach rate constants from 1 up to 106 M-1s-1 with different types of dienophiles (Fig. 4) [3].
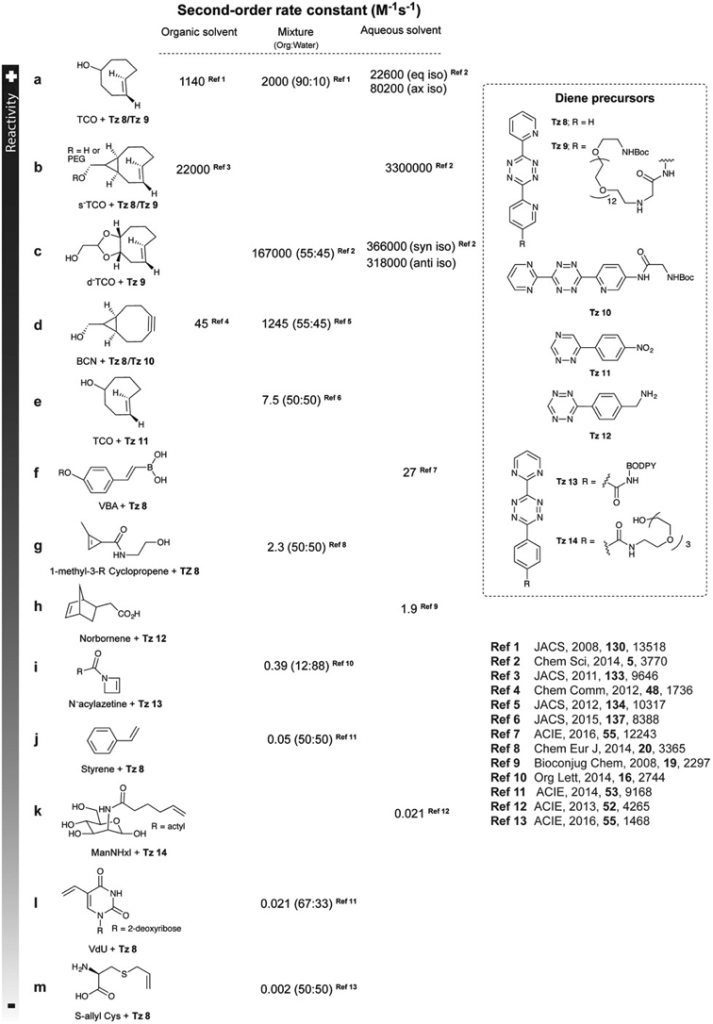
Tetrazines have become important for bioconjugation strategies also due to their high fluorogenic. Since tetrazines are chromophores absorbing light at around 500–550 nm they can act as quenchers towards a series of fluorophores by Förster resonance energy transfer (FRET) and through-bond energy transfer (TEBT). Through IEDDA reaction between the tetrazine and TCO the structure of the tetrazine will be broken which can turn on the fluorescent moiety. Besides, inspired by previous “click-to-release” strategies of the reaction between tetrazine and vinyl ether, The Devaraj group has developed a tetrazine near-infrared fluorogenic probe through a different quenching mechanism: internal charge transfer (ICT) process (Fig. 5) [4].
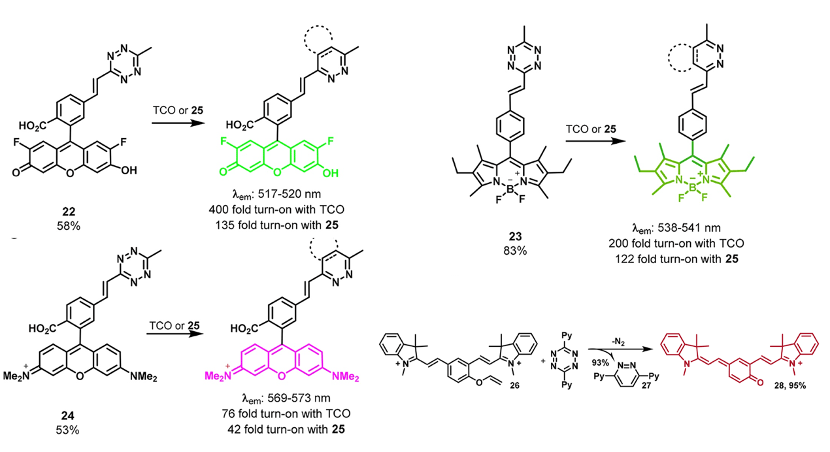
Also, the the aerial oxidation of dihydrotetrazines can be efficiently catalyzed by nanomolar levels of horseradish peroxidase under peroxide-free conditions. The dihydrotetrazine/tetrazine pair can be transferred under the GSH or LED light and photocatalyst. Using this feature Fox group have developed a seirs interesting and useful applications in living cell imaging by light control (Fig. 6) [5].
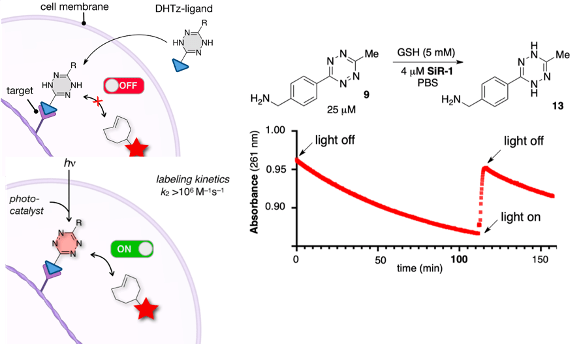
In summary, tetrazine based IEDDA reaction match the principle of the bioorthognoal reactions perfectly. It has high efficiency and can undergo more chemical reactions due to its unique structure compared with the traditional click reaction between azide and alkynes. In resume, the potential of IEDDA reactions is enormous and suggest a rich future of this bioorthogonal reaction in the field of chemical biology.
Reference
- Ellen M. Sletten and Carolyn R. Bertozzi, Angew. Chem. Int. Ed. 2009, 48, 6974 – 6998.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev. 2017, 46, 4895 – 4950.
- Haoxing Wu, and Neal K. Devaraj, Acc. Chem. Res. 2018, 51, 1249−1259.
- Joseph M. Fox, et al, J. Am. Chem. Soc. 2016, 138, 5978−5983.
Evolution of Directed Evolution Methodology
Liu Miao, 8 Dec. 2021
In 1859, Darwin’s On the Origin of Species founded one of the most important theory in biology, the theory of evolution. Although elution theory itself has experienced an evolutionary process, the basic concept of variation–selection–retention cycle unchanged. Adopted from the evolution theory, evolving proteins by mutagenesis and selection was proposed, and in 1993, Arnold and co-workers turned this concept into practice. Since then directed evolution has been widely used to obtain powerful biomolecules. The general procedure of directed evolution includes two main steps: gene diversification and screening/selection (Figure 1). The improved variant will serve as a new starting point for the next round of gene diversification to form a cycle. With the development of chemistry, biochemistry and molecular biology, strategies used in directed evolution have also been evolved.
1. Development of Gene Diversification
In molecular evolution, gene mutation and recombination are two major forces for diversification. For creating gene mutation, random mutagenesis by error-prone PCR was firstly used as the experiment is convenient to carry out, but high-throughput screening methods are necessary because of its random nature. With more knowledge of the protein structure–function relationships revealed by biochemist, focused mutagenesis like site saturation mutagenesis can be performed on critical residues of enzymes, thus creating a smaller but more efficient library. For gene recombination, various DNA shuffling methods were utilized as more molecular biology tools has been developed to imitate the natural process of homologous recombination.
Beside create mutants in test tube, which is labor and time intensive, in vivo gene diversification methods were also developed as more in vivo gene edit tools were found. Early mutation applications in microorganism use E. coli carrying with gene mutation enzyme plasmid, but their applications were limited since they introduce deleterious mutations in the host genome and result in genetic instability. So the development of in vivo gene mutation focused on finding orthogonal mutator plasmids and strains. For in vivo gene recombination, molecular biology tools like bacteriophage λ Red system and Cre-LoxP system were applied. In recent years, CRISPR-Cas system was proved to be a powerful gene edit tool in prokaryotic and eukaryotic cells, which was also used to introduce gene diversifications in vivo.
2. Development of Screening/Selection
Different from creating gene diversification, which is simply relied on the evolution of molecular biology, development of screening/selection methods were functional-oriented and diversified. Whatever the detection method using, the bottle-neck problem is to improve the throughput and efficiency.
For screening methods, each library variant will be assayed individually by using biochemical or biophysical analytical methods to evaluate the desired property. Traditional analytical methods like microplate or chromatography limit with throughputs, thus digital imaging, droplet microfluidics and fluorescence activated cell sorting were applied to increase analytical throughput. For selection methods, the protein function will be linked to the growth or survival of the host organism. The nonfunctional variants will be automatically eliminated during the selection process. So building a general platform for identifying desired phenotypes efficiently is the key challenge for selection methods, solutions include kinds of display-based methods and functional compartments.
3. Speed-up Direct Evolution Process
Beside the evolution on individual step, efforts have also been made to expedite the whole diversification–selection–amplification cycle. First, the efficiency of directed evolution can be greatly influenced by the quality of the library. At the early stage, most directed evolution experiments relied on random diversification, limiting the library quality. To overcome these limitations, rational design can be introduced to reduce the size of the library and simplify screening. With the increase of directed evolution experiments, a giant experimental database can be generate to link the sequence information with functional information, computational tools have been designed to identify promising initial variants. Moreover, in recent years, artificial intelligence has been introduced, structure prediction program like AlphaFold developed by DeepMind has successfully predicted protein structure, which may lead another evolution in this area.
As directed evolution usually need several iterative cycles to get desired variants, the connection between selection rounds is also a point to be optimized. For example, there are hundreds of data obtained in one round using screening method, while in traditional methods only the most efficient one was picked and others were wasted. With the rapid development of data science, machine learning has been applied to solve the problem, which using all the screening data to obtain a sequence-to-function landscape and guide the next round more efficiently. Another example is continuous evolution in vivo. As mentioned before, both gene diversification and selection can be carried out inside the cell, if two parts can be assembled in one compartment without affect the phenotype, the whole directed evolution process can be automated accomplished. Phage-assisted continuous evolution (PACE) is one of the most successful examples.
References
- Wang, Y. et al. Chem. Rev. 2021, 121, 20, 12384–12444
- Packer, M. and Liu, D. Nat. Rev. Genet. 2015, 16, 379–394
- Yang, K.K. et al. Nat. Methods 2019, 16, 687–694
Photocatalytic protein reactions
Bao Yishu, 15 Dec. 2021
Light has been wildly used in chemistry for decades. It can promote bond cleavage, such as azobisisobutyronitrile (AIBN)1, and enable thermally-prohibited transformations by promoting alkenes to their excited states, such as cycloadditions reactions.2 Molecules that interact with visible light can act as mediators for chemical transformations.
Photocatalysts and substrates with an appropriately matched redox potential can generate radical species via visible light and subsequently perform chemical transformations (Fig. 1).3 Photoredox catalysis absorbs light and then undergoes electronic excitation. Substrates with a higher redox potential can accept an electron from the excited photocatalyst, while substrates with a lower redox potential can lose an electron to the excited photocatalyst. In this way, organic radicals are formed on the substrates, which can then undergo further transformation. Subsequently, electrons are returned to, or taken from, the intermediate catalyst species to regenerate the ground state catalyst and participate in the next catalytic cycle.
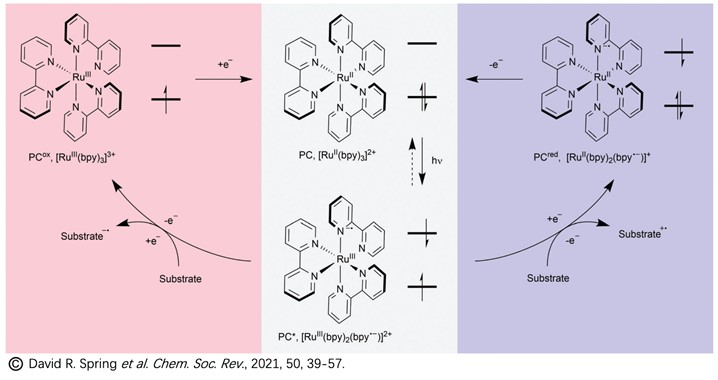
Recent years have witnessed significant progress in photocatalytic reactions for amino acids, peptides, and proteins. For example, MacMillan’s group recently reported a single-site-selective tyrosine modification via photoredox catalysis.4 Owing to the amphiphilicity of phenol, tyrosines often exist in various microenvironments, resulting in different reactivity. Surface-exposed or hydrogen-bond-donating tyrosines are more reactive, while those sterically hindered or engaged in cation-π interactions are deactivated. Based on this microenvironment, the authors designed a bifunctional linchpin molecule that could enable endogenous protein labeling and be suitable for biorthogonal synthesis applications thereafter.
Besides tyrosine, photocatalytic methods are also widely used in cysteine modification. Thiol-ene reactions can be achieved via visible light photocatalysis. In 2017, Wang and co-workers used allyl alcohols and amides as reagents for cysteine and thiosugar modification.5 In 2018, Molander and co-workers developed a method for cysteine arylation using a ruthenium photocatalyst, nickel co-catalyst.6 Protein could also be modified. Wilson and co-workers demonstrated photocatalytic proximity labeling of a native cysteine-containing protein, human MCL-1, using a ruthenium-bipyridyl-modified peptide known to bind MCL-1 in high affinity.7
Compared with previous methods, one advantage associated with photocatalysis is C–H functionalization. Aliphatic residues such as leucine, isoleucine, and glycine, have remained underexploited in amino acid modification due to their inherently inert side chains precluding modifications using traditional methods. Palladium-catalyzed C–H activation is one of the main methods used to target these residues, but many procedures involve biologically-incompatible conditions, such as high temperatures. In contrast, photocatalytic strategies are more suitable for bioconjugation purposes because of the milder conditions.
In summary, photocatalysis shows great promise for amino acids, peptides, and protein modification. Especially, the emergence of C–H functionalization protocols compatible with peptide substrates and capable of modifying aliphatic side chains open a new door for the future of amino acid modification. Despite these great advances, significant challenges remain. Many of the exciting strategies are less effective when applied to larger peptide substrates. In addition, many of the synthesis procedures involve organic solvents, which greatly limits the biological application. For these strategies to be more widely used, biologically compatible reaction conditions need to be further explored.
References
- D. H. R. Barton, D. Crich, W. B. Motherwell, J. Chem. Soc., Chem. Commun. 1983, 939-941.
- K. Singh, W. Trinh, R. Latifi, J. D. Weaver, Org. Biomol. Chem. 2019, 17, 1854-1861.
- T. A. King, K. J. Mandrup, S. J. Walsh, D. R. Spring, Chem. Soc. Rev. 2021, 50, 39-57.
- B. X. Li, D. K. Kim, S. Bloom, R.Y. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes, D. W. C. MacMillan, Nat. Chem. 2021, 13, 902-908.
- G. Zhao, S. Kaur, T. Wang, Org. Lett. 2017, 19, 3291-3294.
- B. A. Vara, X. Li, S. Berritt, C. R. Walters, E. J. Petersson, G. A. Molander, Chem. Sci. 2018, 9, 336-344.
- H. A. Beard, J. R. Hauser, M. Walko, R. M. George, A. J. Wilson, R. S. Bon, Chem. Commun. 2019, 2, 133-142.
Proximal labeling of interacting proteins
CHEN Hongfei, 17 Nov. 2021
Protein-protein interactions (PPIs) play a very important role in biochemical process. Studying PPIs is always a hot topic for many researchers in recent years. Many different approaches such as phage display, yeast hybridization, etc. were developed to illustrate the PPIs, and disclose the composition and organization of protein complexes. However, none of these approaches are based on the real-time and in vivo PPI analysis. Proximity-dependent labeling (PDL) of interacting proteins has been proposed by taking advantage of several enzymes, which can attach the known reactive groups to the nearby proteins covalently.
The mechanism of PDL is to generate a free reactive molecule which can conjugate to nearby proteins during its diffusion process. As shown in the figure (Fig. 1), the protein of interest (bait) is genetically fused to a proximity-based labeling enzyme. The fused enzyme will activate and then release reactive substrates to label proximal proteins. Interacting proteins that are near the bait are more likely to be labeled by the proximity enzyme. Then the quantitative mass spectrometry was used to analyze specific binding partners from background labeling.
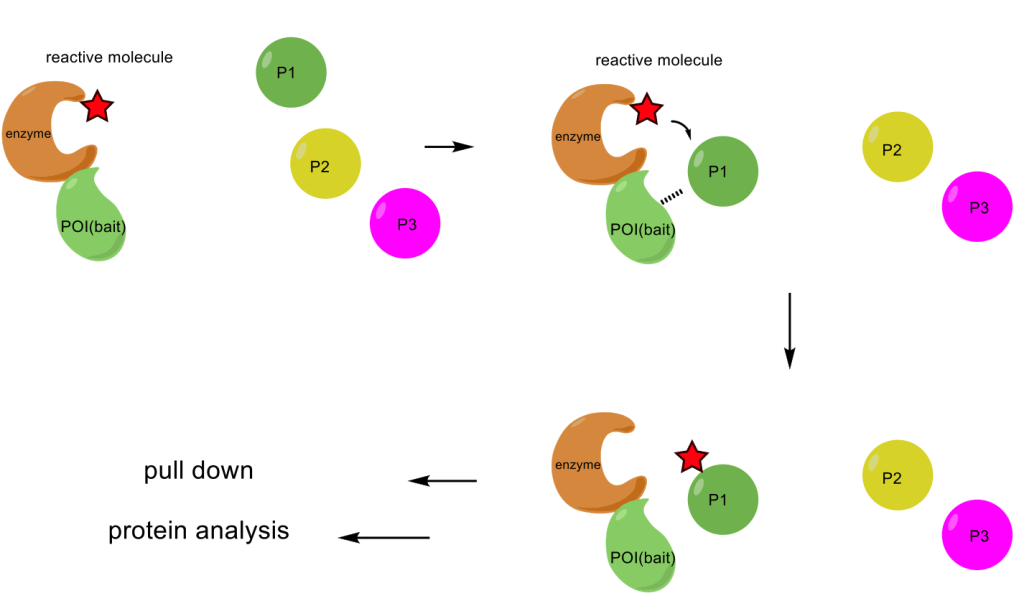
Recently, proximity-tagging systems such as BioID1, EMARS2 and APEX3 have been used to identify PPIs. These proximity labeling enzymes—mostly engineered ligase or peroxidase—have different tagging radii and work well in confined compartments. They are very simple and the output information is rich. Just by using more bait proteins or combining with other traditional methods such as the immune precipitation, PDL can be very useful in identifying novel PPIs. Nowadays, these methods have been applied in studying diseases proteins such as Gag protein in HIV4 and LMP1 protein in EBV5. It is undoubtedly that PDL will have a broader application in studying biological processes, diseases, and therapies.
References
[1] E. de Boer, P. Rodriguez, E. Bonte, J. Krijgsveld et al. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 7480
[2] N. Kotani, J. Gu, T. Isaji, K. Udaka et al. Biochemical visualization of cell surface molecular clustering in living cells. Proc. Natl. Acad. Sci. USA 2008, 105, 7405
[3] Hung, V. et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 (2014).
[4] N. M. Bell, A. M. Lever et al. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136.
[5] K. Holthusen, P. Talaty, D. N., Jr. Everly et al.Regulation of Latent Membrane Protein 1 Signaling through Interaction with Cytoskeletal Proteins. J. Virol. 2015, 89, 7277.
Cell-cell interaction in microorganisms
LIU Min, 10 Nov. 2021
Quorum sensing was first discovered in Vibrio fischeri by Nealson and colleagues in 1970. They observed that when the bacteria density is low, no fluorescence could be seen. But when the bacteria density reaches a high level, it will show fluorescence. This phenomenon is related to the expression of a fluorescent gene in a bacterium, which is only expressed under the induction of autoinducer. Therefore, bacteria with low population density will not express relevant genes. Autoinducer is a compound that can regulate the expression of specific genes in bacteria. Many autoinducers discovered were derived from molecules with AHL as the backbone. Later, scientists also discovered some oligopeptide autoinducers and other autoinducers with special structures. Under natural conditions, some bacterial cells can gather cooperator and cheater, and the protease secreted by cooperator can enter the symbiotic environment to promote the growth of cheater. In addition, the quorum sensing of Vibrio cholerae is also very common. When Vibrio cholerae exists in the ocean, the density of bacteria is relatively low, so the quorum sensing effect is very weak, which is conducive to the formation of biofilms. When the bacteria enter the host, the density of the bacteria increases and the quorum sensing increases, so the formation of biofilms is inhibited, which is the scenario of vibrio cholerae in the human body.

In synthetic biology, synthetic consortia, a group of bacterial flora, can be used to synthesize natural products, showing advantages of high efficiency, stability, controllability. For example, the Gregory group used the E. coli and yeast multi-host system to produce the precursor of paclitaxel. Besides, the synthetic co-culture controller constructed by the Bentley group could control the growth of bacteria.
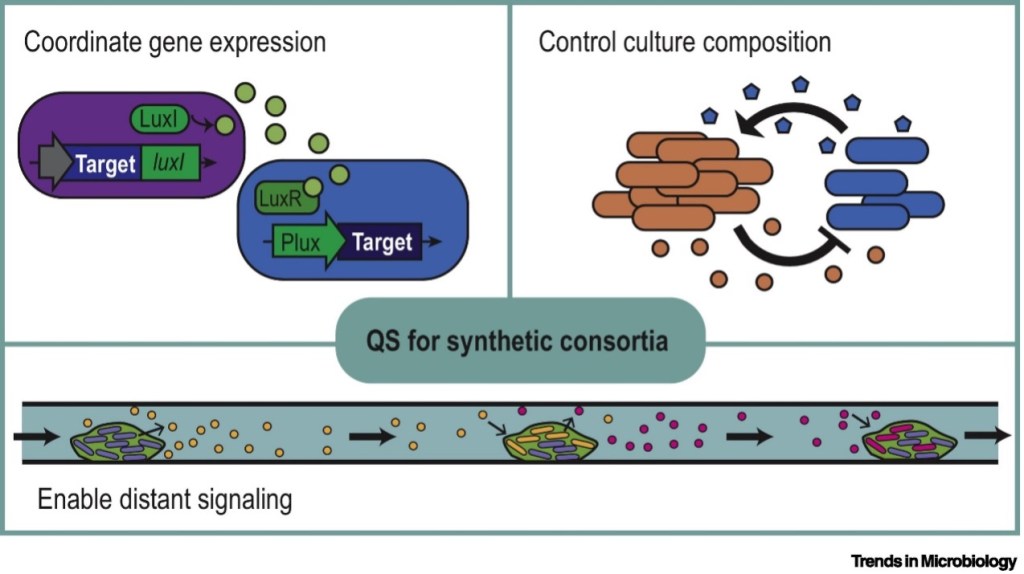
In summary, by studying the interaction between microorganisms, we can realize the process from discovery to application. The group effect is still a treasure that the current research can focus on.
References:
Pérez-Velázquez, Judith, Meltem Gölgeli, and Rodolfo García-Contreras. “Mathematical modelling of bacterial quorum sensing: a review.” Bulletin of mathematical biology 78.8 (2016): 1585-1639.
Schaefer, Amy L., et al. “Quorum sensing in Vibrio fischeri: probing autoinducer-LuxR interactions with autoinducer analogs.” Journal of bacteriology 178.10 (1996): 2897-2901.
Stephens, Kristina, and William E. Bentley. “Synthetic biology for manipulating quorum sensing in microbial consortia.” Trends in microbiology 28.8 (2020): 633-643.
Zhou, Kang, et al. “Distributing a metabolic pathway among a microbial consortium enhances production of natural products.” Nature biotechnology 33.4 (2015): 377-383.
Stephens, Kristina, et al. “Bacterial co-culture with cell signaling translator and growth controller modules for autonomously regulated culture composition.” Nature communications 10.1 (2019): 1-11.
Kelly, Robert C., et al. “The Vibrio cholerae quorum-sensing autoinducer CAI-1: analysis of the biosynthetic enzyme CqsA.” Nature chemical biology 5.12 (2009): 891-895.
Synthetic Biology: A glance into the future
Wang Yue, 3 Nov. 2021
It happens that synthetic biology is poised to improve our health in myriads of ways. For example, synthetic biology can produce renewable fuels and manufactured goods, and assist human health in terms of pharmaceutics, nutraceuticals and even therapeutics.
“What I cannot create, I do not understand ”, is a famous saying quoted from Richard Feynman. It is also the essential idea of researchers of synthetic life. In May 2010, John Craig Venter published an article on science[1] describing their research of successfully creating “synthetic cell”. This was accomplished by synthesizing a very long DNA molecule containing an entire mycoplasma genome, and introducing it into another cell. This news has caused a sensation at that time, and people believed that the era of synthetic life will arrive in the near future.
At the same time, Jef Boeke, professor of New York University, was spearheading an ambitious effort to design and build an entire yeast genome from scratch, known as the Sc2.0 project[2].

As a eukaryote, a category that includes humans and other animals, S. cerevisiae has a more complex genome than mycoplasma. So Jef formed an international consortium to create a synthetic version of the full S. cerevisiae genome within 5 years. Chinese scientists from Tianjin University, Tsinghua University and BGI-Shenzhen have taken part of the work in this project and so far they have assembled four synthetic yeast chromosomes, making China the second country capable of designing and building eukaryotic genomes.
In 2016, researchers led by Craig Venter at the J. Craig Venter Institute in San Diego, California, announced that they had created synthetic “minimal” cells. The genome in each cell contained just 473 key genes thought to be essential for life. The cells were

named JCVI-syn3.0 after the institute and they were able to grow and divide on agar to produce clusters of cells called colonies. But on closer inspection of the dividing cells at the time, Venter and his colleagues noticed that they weren’t splitting uniformly and produce daughter cells of unnatural shapes and sizes. By reintroducing various genes into these synthetic bacterial cells and then monitoring how the additions affected cell growth under a microscope, Strychalski pinpoint seven additional genes required to make the cells divide uniformly. After adding 5 more genes into the JCVI-syn3.0, synthetic cells eventually can grow and divide into cells of uniform shape and size, just like most natural bacterial cells[3].

Gene therapy
Synthetic biology also make the medicines of tomorrow look different from those of today by editing the existing bacteria in vivo or delivering modified bacteria to our bodies.
For example, Eligo Bioscience, based in Paris, is a company that combines gene editing and microbiota modification. Their founders, Dr. Duportet pointed out that in the past few years, there have been multiple validated examples showing the expression of specific bacterial genes and diseases. Eligo’s goal is to use a CRISPR-based system to inactivate these genes-there is a proven link between bacterial genes and disease. Eligo’s drug “Eligobiotics” consists of a viral vector (a phage particle without a phage genome) that contains a gene editing payload and is specific to the target bacteria. Once the CRISPR system is delivered, editing takes place in the body and in the intestines, inactivating genes and destroying bacteria. One candidate for Eligo is addressing antimicrobial resistance (AMR) bacteria in a project supported by the non-profit organization CARB-X. Eligobiotic, known as EB004, aims to remove the AMR gene from the bacteria in the intestines of patients undergoing organ transplantation to prevent bacterial infections. A transplant patient with AMR bacteria in the intestines has a 50% chance of contracting AMR bacteria after transplantation. In the case of infection, the mortality rate jumped to 70%. If AMR bacteria are removed, the mortality rate will drop to 4%.
Based on synthetic biological technology, Synbiotics, a Chinese company based in Beijing, has designed and developed a gene circuit that can accurately identify tumors and improve the killing effect, and insert the gene circuit into the adenovirus vector, forming a method that can accurately identify tumors, improve the immune environment, and effectively improve SynOV system, a new oncolytic virus gene therapy drug platform with tumor killing ability.
Surprisingly, on November 27, 2020, Synbiotics announced that its first gene therapy product, SynOV1.1, developed based on domestically original synthetic biotechnology, has been approved by the US FDA for clinical trials for the treatment of alpha-fetoprotein (AFP), including advanced liver cancer. ) Positive solid tumors, and plans to carry out Phase I/IIa clinical studies at the Memorial Sloan Kettering Cancer Center in the United States in the near future. The application for clinical trials of this drug in China is also underway.
Metabolic Engineering
Metabolic engineering is a multidisciplinary engineering toolbox for modern production in daily life. In which,Jay Keasling’s team takes the leading position. In the 1970s, Chinese scientists rediscovered it and identified its active ingredient, artemisinin, and artemisinin is now extracted from sweet wormwood grown commercially in China, Southeast Asia and Africa. However,The quality, supply and cost have been unpredictable and inconsistent.
In 2006, Jay reported to synthesize small amounts of artemisinic acid, which is chemically analogous to the actual drug. Using synthetic biology techniques from Keasling’s lab, Amyris added that gene to yeast along with other plant genes to boost artemisinic acid production by a factor of 15, good enough to interest Sanofi.
Then Keasling has turned to cannabis, where his work endows brewer’s yeast with the capacity to make cannabinoids. Keasling first published a paper on Nature back in 2019[4], but at this point, it is unclear if the process could be scaled up enough to support a new industry.
Reference:
[1] Gibson D G, Glass J I, Lartigue C, et al. Creation of a bacterial cell controlled by a chemically synthesized genome[J]. Science, 2010, 329(5987): 52-56.
[2] Annaluru N, Muller H, Mitchell L A, et al. Total synthesis of a functional designer eukaryotic chromosome[J]. Science, 2014, 344(6179): 55-58.
[3] Pelletier J F, Sun L, Wise K S, et al. Genetic requirements for cell division in a genomically minimal cell[J]. Cell, 2021, 184(9): 2430-2440. e16.
[4] Luo X, Reiter M A, d’Espaux L, et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast[J]. Nature, 2019, 567(7746): 123-126.
